
锆合金氧化膜中氢吸附行为的第一性原理研究
2021-09-25 11:54:10谢耀平胡丽娟
上海金属 2021年5期
李 彤 谢耀平 胡丽娟 王 洋
(1.上海大学材料研究所,上海200072;2.上海大学微结构重点实验室,上海 200444;3.上海大学计算机学院高性能计算中心,上海 200444)
锆的热中子吸收截面小,在其基础上添加少量合金元素制得的锆合金具有良好的力学性能和优异的高温耐腐蚀性能,因而被广泛用作压水堆核电站中核燃料元件的包壳材料[1]。锆合金的耐水侧腐蚀性能是影响包壳寿命的关键因素[2-3],而锆合金氧化膜对其耐腐蚀性能有重要的影响。另一方面,在核反应堆中运行时,锆合金包壳氧化并和水蒸气反应产生氢气。因此,研究锆合金氧化膜的氢吸附机制,对理解锆合金氧化膜性质,揭示锆合金的腐蚀机制有重要意义。
为了揭示氧化膜中氢的行为,Sundell等[4]利用原子探针层析成像技术,研究了在H2O和D2O中进行高压釜测试的锆合金氧化膜中氢和氘的分布,结果表明,腐蚀过程中产生的氢进入锆合金氧化膜中,且容易在氧化膜晶界偏聚;另一方面,通过原子尺度计算也可以理解氧化膜中氢的行为。目前,研究人员对氧化锆与氢之间的相互作用开展了一些新的研究。例如Syzgantseva等[5]通过第一性原理计算研究了单斜氧化锆与H2之间的相互作用,利用平板模型构建了(1-11)和(1-01)ZrO2表面,发现氢分子在ZrO2表面的吸附为物理吸附,氢原子在该表面的吸附能较大,属于化学吸附,且氢原子的吸附引起了ZrO2表面重构。Jin等[6]利用第一性原理计算研究了(ZrO2)n(n=1~6)团簇上氢分子的吸附行为及其电子结构,发现氢分子容易被(ZrO2)n(n=1~6)团簇吸附,且在ZrnO2nH2(n=1~6)簇中,Zr5O10H2簇的结合能最低;通过计算振动频率和Mulliken电荷,发现在不同大小的ZrnO2nH2团簇中都形成了H—O和Zr—H键;通过分析电荷密度分布可知,吸附后电荷从(ZrO2)n团簇转移到H2上,这是团簇与氢分子之间的电子轨道杂化引起的。此外,还发现随着(ZrO2)n团簇上吸附的H2分子数目的增加,吸附逐渐倾向于向低配位数的原子位置迁移。
由于锆合金氧化膜性质对环境介质与锆合金基体之间的元素传输起着决定性的作用,研究氧化膜中氢吸附行为具有重要的意义。本文从原子层次上揭示了氢与氧化膜的相互作用,以及氢在氧化膜中的存在形式与状态。由于锆合金氧化膜主要由单斜相ZrO2组成,晶界是腐蚀过程中离子输运的重要通道,采用第一性原理计算方法研究了单斜氧化锆中氢吸附行为,并重点分析了氢在晶界的吸附行为。
1 计算方法与模型构建
1.1 模型构建
采用优化的ZrO2{210}/[001]26.29°倾斜晶界计算氢的吸附行为,该晶界具有较高的对称性,超级原胞中的原子数相对较少[7-8]。采用平板模型构建该晶界,如图1(a)所示。虚线部分是晶界位置,为了避免结构中出现多个晶界,模型引入了真空层。为了消除超胞大小不同引起的计算误差,m-ZrO2晶内的理想晶体结构也采用了与晶界超胞尺寸相似的超级原胞,如图1(b)所示。然后分别研究晶界或晶内不同位置氢的吸附,吸附位点如图1(c,d)所示。
1.2 计算方法
本文所涉及的能量计算及电子结构计算均采用基于密度泛函理论(density functional theory,DFT)的第一性原理计算方法(first-principles method),具体通过VASP(Viennaab-initio simulation package)软件包实现[9]。离子与价电子之间的相互作用采用投影子缀加波(projector augmented wave,PAW)来描述[10-11],电子与电子之间的交换关联泛函势采用广义梯度近似(generalized gradient approximation,GGA)下的Perdew-Burke-Ernzerhof(PBE)泛函表示。平面波的截断能均为400 eV,k点网络采用Monkhorst-Pack方法生成[12],对于模拟构建的晶体结构,超胞k点网格均设置为(2×2×1)。计算时对晶胞内所有原子的位置都进行了充分的弛豫,其能量的收敛标准为0.01 eV/Å。
氧化膜晶界和晶内氢吸附能计算公式分别为:
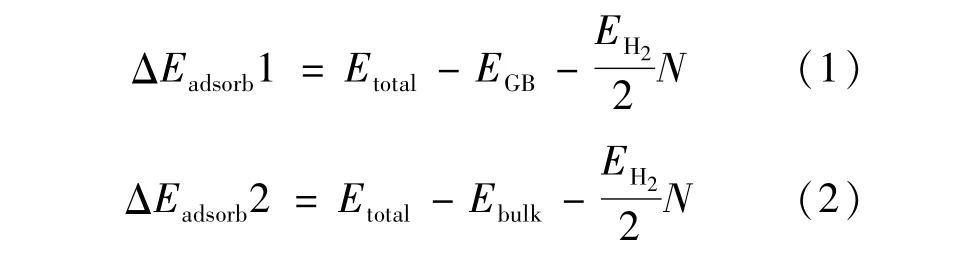
式中:Etotal为吸附氢原子或氢分子的ZrO2晶界(晶体)结构的总能;EGB为纯净晶界结构的总能;Ebulk为纯净晶体结构的总能;EH2为氢分子的总能,N为体系中吸附的氢原子总数(包括以分子和原子形式吸附的两种形态)。
2 计算结果与讨论
2.1 氢原子吸附能的计算
如图1所示,在m-ZrO2晶界选取21个位置添加氢原子。表1为晶界氢原子吸附位点与最邻近锆原子之间的距离。表2为单个氢原子在图1(c)中不同吸附位置的吸附能。从表2可以看出,ZrO2晶界不同位置氢原子的吸附能不同,在-4.39~ +3.43 eV/atom之间。当吸附能为负值时,表明该位置氢的吸附为放热过程,吸附容易发生。反之,当吸附能为正值时,该位置氢的吸附难以发生。结合图1和表2可以发现,吸附位点处“间隙”越大,吸附能越小。
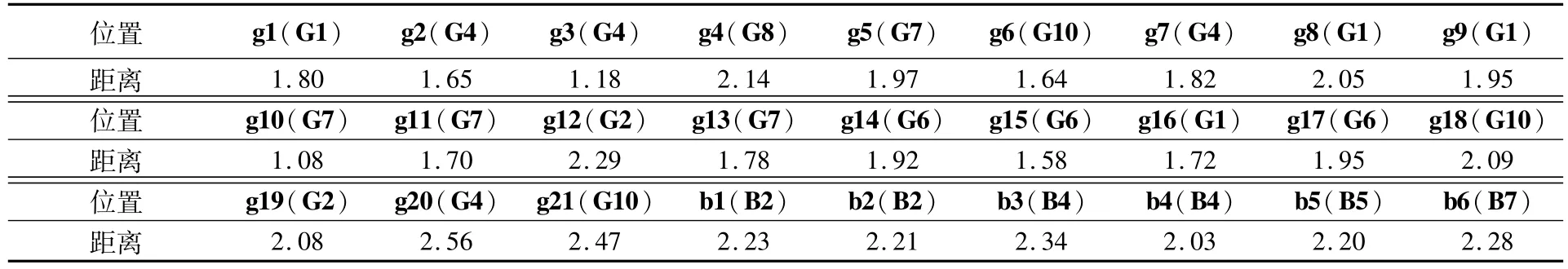
表1 氢原子吸附位点与最邻近锆原子之间的距离Table 1 Distance between hydrogen adsorption site and the nearest zirconium atomÅ

表2 m-ZrO2晶界单个氢原子的吸附能Table 2 Adsorption energy of a single hydrogen atom in m-ZrO2grain boundary
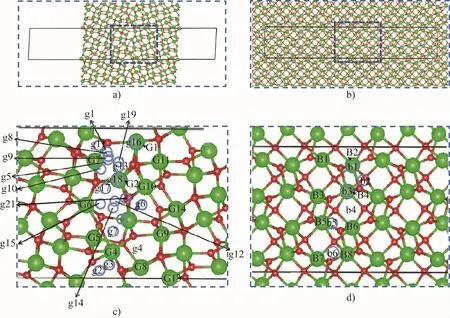
图1 m-ZrO2原子结构((a,b)为理想m-ZrO2晶粒和含有晶界的m-ZrO2超级原胞内部;(c,d)分别为(a,b)中蓝色虚线中原子结构放大图。黑色实线代表超晶胞的边界,深蓝色虚线框为计算模型中的原子吸附区域,绿球代表Zr原子,红球代表O原子,蓝圈代表吸附氢原子的位置,蓝圈上bm和gm(m=1,2…21)分别代表晶粒内部与晶界处典型的原子吸附位点,吸附能小于0的用蓝色标记,大于0的用黑色标记。Gm(m=1,2…21)代表晶界Zr原子,Bm(m=1,2…8)代表晶内Zr原子)Fig.1 Atomic structures of m-ZrO2((a,b)are the interior of the ideal m-ZrO2grains and the m-ZrO2 supercells containing grain boundaries;(c,d)corresponding to the enlarged atomic structures in the blue dotted line in(a,b),respectively.Solid black line indicates the boundary of the supercell,dark blue dotted frame marks the atom adsorption area in the calculation model,green ball indicates the Zr atom,red ball indicates the O atom,blue circle indicates the position where hydrogen atom is added,bm and gm(m=1,2…21)represent typical atomic adsorption sites inside the grain and at the grain boundary,respectively,those with adsorption energy less than 0 are marked with blue and those with adsorption energy greater than 0 are marked with black.Gm(m=1,2…21)represents Zr atoms at the grain boundary,Bm(m=1,2…8)marks the Zr atom inside the grain)
对氢原子在m-ZrO2完美晶体内的吸附进行了研究,对晶内8个不同的锆原子进行标记,根据对称性选择了6个典型的氢吸附位置,如图1(d)所示。晶内氢原子吸附位点与最邻近锆原子之间的距离也列于表1,吸附能的计算结果如表3所示。可以看出,吸附能均为正值,最低2.00 eV左右,说明氧化膜晶内难以吸附氢原子,氢原子更易吸附在氧化膜的晶界,这与Sundell等[4]的试验结果一致。由于晶界在材料中体积比很小,而且晶粒内部对氢的吸附较弱,因此氧化膜总体吸氢能力不强[13]。虽然氢的吸附量不大,但晶界的氢吸附会导致晶界力学性能变化,引起氧化膜中微孔隙和微裂纹形成难易程度的变化,是影响腐蚀的重要因素。

表3 m-ZrO2完美晶体内单个氢原子的吸附能Table 3 Adsorption energy of a single hydrogen atom in the perfect m-ZrO2lattice grain
接着研究了多个氢原子在晶界的吸附,计算了超胞中含有2~4个氢原子时的吸附性质。2个氢原子以原子或分子形式吸附。选取典型的吸附位置以研究吸附能随超胞中氢原子数量的增加而变化的情况。对于2个氢原子吸附:选取单个氢原子吸附能最低的吸附点进行组合,如g1+g2;还选取一些分散的吸附位点进行组合,如g1+g5和g5+g19。由于氢原子在g5位置的吸附能较低,氢分子均吸附在g5位置。对于3~4个氢原子的吸附也采用同样的方法进行组合。将计算得到的1~4个氢原子在晶界的吸附能列于表4。发现含有4个氢原子的超胞中,吸附能仍为负值,表明这些吸附均为放热反应。随着氢原子数量的增加,吸附能的绝对值减小。分析可知,由于晶界空间有限,随着氢原子数量的增加,其电子之间的泡利排斥力增大,导致系统总能提高,吸附能接近于零,然后出现正值。据此可粗略推断,当氢原子数量达到5个后,吸附能将变为正值,吸附转变为吸热反应,吸附更困难。另外还发现,氢分子或氢原子吸附的吸附能均可能为负值,说明晶界氢的吸附既能以原子形式存在,又能以分子形式存在。

表4 m-ZrO2晶界多个氢原子的吸附能(粗体表示一定数量氢原子情况下最低吸附能)Table 4 Adsorption energy of multiple hydrogen atoms at grain boundary of the m-ZrO2(bold figure indicates the lowest adsorption energy for a certain number of hydrogen atoms)
2.2 氢原子吸附机制分析
晶界不同位置氢原子的吸附能差异较大,为此对晶界氢原子的吸附机制进行分析。单斜ZrO2中Zr原子配位数为7,O原子配位数为3或4。氢在Zr原子上吸附较为稳定[6],因此考察了ZrO2{210}/[001]26.29°晶界Zr原子的配位数,结果列于表5。从表5可以看出,晶界出现了配位数为5或6的Zr原子,这些Zr原子上将形成悬挂键[14-16]。低配位数的Zr原子上氢的吸附能可能更低。
结合表5配位数和表2吸附能发现,氢原子吸附能最低的位置出现在配位数较低的Zr原子周围。例如,g1位置的氢吸附能最低,其附近存在一个配位数最低的Zr原子G7(配位数为5);g2、g3位置的吸附能也较低,其附近存在一个配位数低的Zr原子G12(配位数为6)。这与上文分析的一致。进一步对氢原子吸附位点附近的Zr原子进行电子结构分析。通过计算态密度可以揭示原子间形成的化学键性质,因此计算了晶界处具有低配位数的Zr原子(G3/4/5/7,配位数分别为7、6、5)的局域态密度,并将6配位、5配位Zr原子分别与7配位Zr原子的态密度进行比较,如图2(a,c,e)所示。从图中可以看出,在价带顶(valence band maximum,VBM)附近,原子配位数越低,态密度值越高。其中5配位Zr原子在靠近价带顶的位置态密度值最高,因此其电子容易发生转移成键,化学性质更活泼,更容易吸附氢,从而解释了低配位Zr原子容易吸附氢的原因。因此,晶界低配位的Zr原子位置更容易吸附氢。
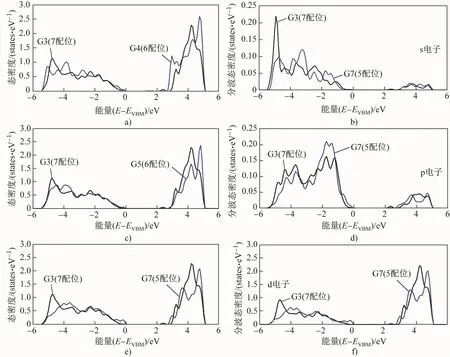
图2 G3、G4、G5和G7锆原子的局域态密度(a,c,e)以及G3、G7锆原子的分波态密度(b,d,f)(EVBM为价带顶)Fig.2 Local density of states for G3,G4,G5 and G7 Zr atoms(a,c,e)as well as projected density of states for G3,G7 Zr atoms(b,d,f)(EVBMis valence band maximum)

表5 m-ZrO2晶界Zr原子的配位数Table 5 Coordination number of Zr atom at grain boundary of the m-ZrO2
G3、G7锆原子的分波态密度如图2(b,d,f)所示。对于s、p、d电子态,5配位Zr原子在价带顶附近的态密度均大于7配位Zr原子的。Zr原子的电子态主要由d电子贡献,s和p电子的贡献较少,说明影响吸附能大小的主要是Zr原子的d能带,这与Hammer等[17]的d-band理论一致。另外,ZrO2中各个吸附位点周围原子结构各有特点,原子的键长、键的取向均不相同,这都会引起电子结构变化,进而导致氢原子的吸附能不同,这是具有相同配位数吸附位点的吸附能有差别的主要原因。
3 结论
锆合金氧化膜晶界比晶内更容易吸附氢,氢以原子或分子形式被晶界吸附。随着氢含量的增加,晶界吸附氢的能力逐渐减弱。当原子的配位键低时,靠近价带顶的电子态增多,化学性质更活泼,因此氢原子更易于吸附在低配位键的原子周围。
猜你喜欢
上海金属(2022年4期)2022-08-03 09:52:00
中国新技术新产品(2022年7期)2022-07-14 11:37:48
工程科学学报(2021年10期)2021-10-23 13:51:40
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09 06:12:08
原子与分子物理学报(2021年2期)2021-03-29 07:31:26
当代陕西(2019年6期)2019-04-17 05:04:10
数理化解题研究(2017年16期)2017-07-21 09:32:29
中学物理·高中(2016年8期)2016-08-08 09:21:06
山西大同大学学报(自然科学版)(2016年6期)2016-01-30 08:29:23
上海金属(2015年6期)2015-11-29 01:09:02
