
氯离子环境中掺杂元素对氧化铝表面腐蚀影响的第一性原理研究
2021-09-25 11:54祖武杰
上海金属 2021年5期
祖武杰 史 文,2 董 瀚,2 王 洋
(1.上海大学材料科学与工程学院,上海 200444;2.上海大学省部共建高品质特殊钢冶金与制备国家重点实验室,上海 200444;3.上海大学计算机工程与科学学院,上海 200444)
铝及铝合金表面生成的致密氧化膜具有良好的耐蚀性,能够有效保护合金基体不受腐蚀。但在含Cl-的海洋或沿海大气环境中,铝合金极易因表面氧化膜被破坏而发生腐蚀[1]。表面处理能有效提高合金氧化膜耐Cl-腐蚀性能,如离子注入合金元素掺杂氧化铝膜、激光表面合金化等方法[2-3]。然而,由于试验情况的复杂性和分析手段的局限性,掺杂元素对氧化铝耐Cl-腐蚀的微观影响机制还不清楚。密度泛函理论(DFT,density functional theory)是一种在原子尺度上研究表面与分子或原子之间相互作用机制的有效工具,通过DFT计算可以从原子尺度上了解Cl原子与氧化铝表面之间的相互作用,如对腐蚀有重要影响的吸附行为等[4-6]。为了解掺杂元素对氧化铝耐Cl-腐蚀性能的影响,本文采用第一性原理计算方法,结合溶剂化处理和电子结构分析,研究了含Ga或Sn的氧化铝表面Cl原子的吸附行为,探讨了Ga和Sn对氧化铝表面耐腐蚀性能的影响及其机制。
1 计算方法与细则
计算采用基于密度泛函理论的VASP(vienna ab-initio simulation package)软件包[7]。使用平面波基组和周期性边界条件求解Kohn-Sham方程,采用广义梯度近似(GGA,generalized gradient approximation)的Perdew-Burke-Ernzerhof(PBE)描述电子的交换关联函数,Ion cores采用PAW(projector augmented wave)描述[8]。布里渊区k点网格采用Monkhorst-Pack方法,以Gamma点为中心进行采样。
对密排六方结构的α-Al2O3晶胞进行优化,平面波截断能取650 eV,k点取15×15×5。优化后的晶格参数为a=b=4.807 8 Å,c=13.123 2 Å,与其他工作[9-10]中的计算值和试验值相近,误差在1%以内。计算采用的α-Al2O3(0001)表面模型为以1层铝原子层为终止面的非极性模型[11],以表面能变化小于0.01 J/m2为收敛标准,设置原子层数为12层,真空层厚度为15 Å,并允许所有原子弛豫,平面波截断能取650 eV,k点取8×8×1。用掺杂原子替换氧化铝表面的1个铝原子空位。考虑了Cl原子在掺杂表面的所有高对称性吸附位点,吸附位点以X(Y)-Z的形式表示,X表示吸附原子,Y表示吸附原子所在层数,Z表示同一层吸附原子的不同位点(Z=1,2,3,…),按照距离掺杂原子由近及远的顺序排列,初始吸附距离为共价键半径的总和。Al2O3(0001)表面原子结构及吸附位点如图1所示,浅蓝色原子为Al原子,红色原子为O原子,紫色原子为掺杂原子X(X=Ga,Sn)。
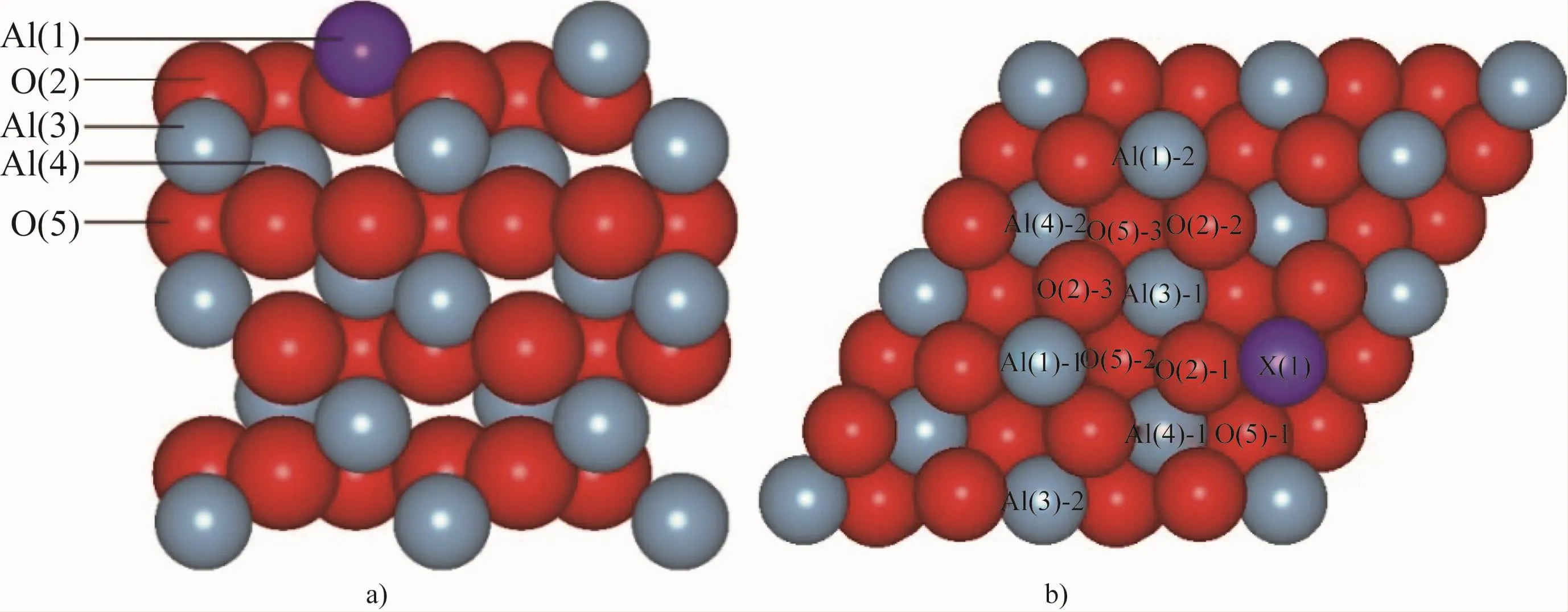
图1 Al2O3(0001)表面原子结构(a)及吸附位点示意图(b)Fig.1 Schematic diagrams of atomic structure(a)and adsorption site(b)of Al2O3(0001)surface
氧化铝的腐蚀通常发生在溶液环境中,溶剂对表面的腐蚀反应有重要影响。因此,为了更有效和精确地模拟Al2O3表面与溶剂之间的固/液界面,采用VASPsol[12-13]进行溶剂化处理,模拟水环境,水的介电常数设定为80。吸附物与表面之间的吸附作用会引起偶极效应[14],尤其是α-Al2O3这类离子性较强的金属氧化物,并且在溶剂化模式中,这种偶极场作用会增大[9]。同时,VASPsol溶剂化处理也可能在表面产生不稳定偶极子[15]。因此,通过设置偶极场补偿,对局部电位进行自我校正,以消除偶极子与周期性模型之间的假性相互作用[16]。
采用表面吸附能Ea来反映不同掺杂表面对Cl原子吸附能力的强弱,计算公式为:
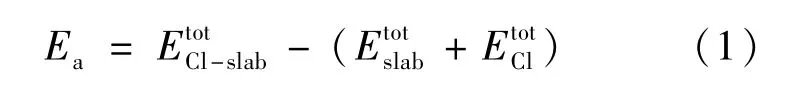
2 结果与讨论
2.1 含Ga或Sn的Al2O3表面Cl原子吸附行为
水环境中Cl在未掺杂Al2O3表面(Nonedoped)不同位点的吸附能和吸附距离计算结果如表1所示,表中d为Cl的吸附距离,即Cl与Al2O3最外表面之间的距离。可以看出,Cl在None-doped表面的吸附位点可能有3个,Al(1)-1位点的吸附能最低,为-3.59 eV,表明Cl吸附在Al(1)-1位点时最稳定,这与Zhang等[17]的研究结果一致,该位点Cl的吸附距离也接近试验测量值[18]。水环境中Cl在含Ga或Sn的Al2O3掺杂表面不同位点的吸附能和吸附距离计算结果也如表1所示。通过比较Cl在Al2O3掺杂表面和未掺杂表面的吸附能,可以得出掺杂元素对氧化铝耐Cl-腐蚀性能的影响。如果Al2O3掺杂表面对Cl的吸附能力下降,则Al2O3掺杂表面吸附Cl后释放的能量更多,吸附过程的驱动力更大,该掺杂元素将促进Cl在Al2O3表面的吸附,反之则相反。根据表1分别讨论掺杂Ga(Ga-doped)和掺杂Sn(Sn-doped)的Al2O3表面Cl的吸附行为。可以看出,Al2O3表面掺杂Ga或Sn后,Cl可能的吸附位点均为4个。对于Ga-doped表面,Cl在Ga(1)位点的吸附能最低;在Sn-doped表面,Cl在Al(1)-1位点的吸附能最低。Ga-doped表面Cl的最低吸附能高于None-doped表面,而Sndoped表面Cl的最低吸附能低于None-doped表面。这表明Ga降低了Al2O3表面对Cl的吸附能力,而Sn增强了Al2O3表面对Cl的吸附能力。

表1 Cl在未掺杂和掺杂Ga或Sn的Al2O3表面不同位点的吸附能和吸附距离Table 1 Adsorption energies and adsorption distances of Cl at different sites on the none-doped and Ga-or Sn-doped Al2O3surfaces
吸附可分为物理吸附和化学吸附。对于材料的腐蚀,如果吸附质在表面是化学吸附,与表面形成化学键,不易脱附,则易与表面产生更多的其他作用。因此,有必要分析Cl在表面的吸附状态,以便更好地了解掺杂元素对Al2O3表面Cl吸附行为的影响。电子局域函数(ELF,electron localization function)可以反映模型中电子的局域化程度,常用来分析原子之间的成键情况。计算了Cl在不同掺杂表面的最稳定吸附结构中各原子的电子局域化分布特征,进而确定Cl与表面之间的相互作用,如图2所示。图2中绿球为Cl原子,蓝球为Al原子,红球为O原子,其他颜色圆球对应为掺杂原子,黄色区域为等势面,ELF=0.855。从图2可以看出,未掺杂和掺杂Al2O3表面的Cl原子与表面原子之间均存在一个明显的电子高度局域化区域,这表明Cl与表面原子之间有明显的成键作用,Cl在Al2O3表面是化学吸附。
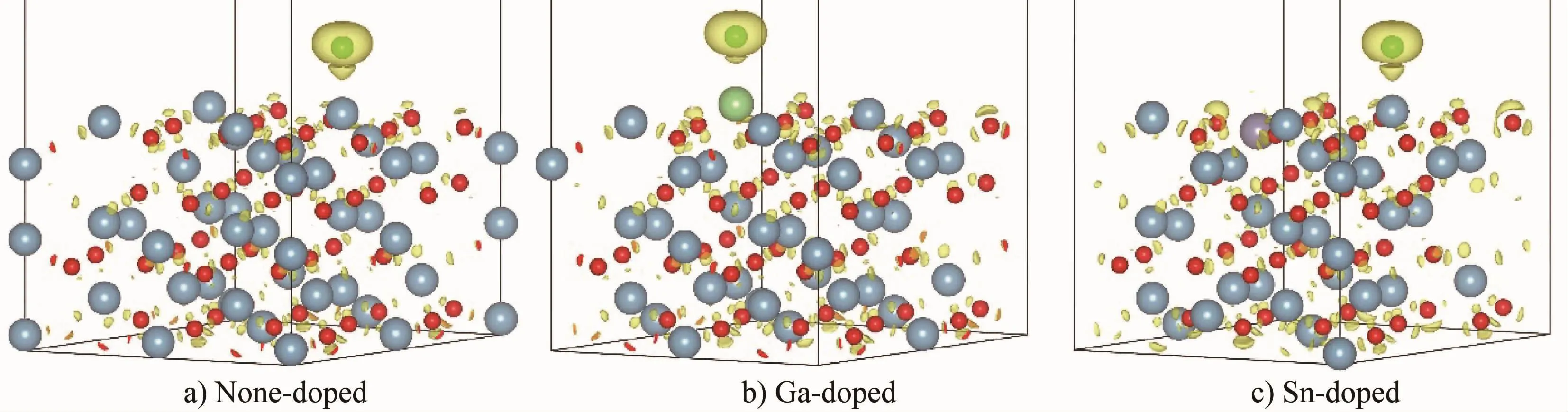
图2 Cl在未掺杂和掺杂Ga或Sn的Al2O3表面的最稳定吸附结构的ELF图Fig.2 ELF maps of the most stable adsorption structure of Cl adsorption on the none-doped and Ga-or Sn-doped Al2O3surfaces
差分电荷密度图可以给出模型中电荷转移的情况,进而了解原子之间的相互作用,计算方法为吸附后的表面模型的电荷密度减去未吸附的表面模型的电荷密度和孤立吸附原子的电荷密度。图3给出了Cl在不同掺杂表面的最稳定吸附结构的差分电荷密度图,浅黄色区域为得电子区,等势面为0.010 e/Bohr3,浅蓝色区域为失电子区,等势面为-0.002 e/Bohr3。从图3(a)可以看到,Cl与Al原子之间有明显的得电子区域,表明O原子周围失电子明显,O原子的电荷转移到Cl和Al之间,促使Cl—Al化学键的形成,使Cl化学吸附在Al2O3表面。在含Ga或Sn的Al2O3表面也存在Cl吸附后引起表面电荷重新分布的现象。图3(b)显示,Cl吸附在Ga原子上后,使Ga及其周围O原子失去电荷,并向Cl原子周围转移,促使Cl与Ga成键。图3(c)显示,在Sn-doped表面,Cl吸附在Al原子上后,Al原子失电荷明显,电荷转移到Cl原子周围,形成了Cl—Al化学键。差分电荷密度图和电荷局域密度图均表明,Cl在未掺杂和掺杂Ga或Sn的Al2O3表面的最稳定吸附状态为化学吸附。
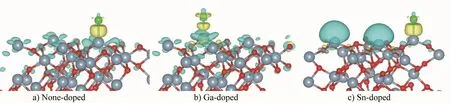
图3 Cl在未掺杂和掺杂Ga或Sn的Al2O3表面的最稳定吸附结构的差分电荷密度图Fig.3 Induced charge density of the most stable adsorption structure of Cl adsorption on the none-doped and Ga-or Sn-doped Al2O3surfaces
2.2 Ga和Sn对Al2O3表面Cl吸附行为的影响机制
原子或分子在合金表面的吸附行为不仅受吸附物性质的影响,也受表面结构的影响。由于各元素性质不同,掺杂后对表面原子电子结构的影响也不同。计算发现,不同类型掺杂表面Al(1)-1位点均有可能吸附Cl原子,且吸附结构相似,因此该位点Cl的吸附行为基本不受表面结构的影响,与表面原子的电子结构的关联性更大。为进一步探究掺杂元素对Al2O3表面电子结构的影响,计算了掺杂不同元素的Al2O3表面Al(1)-1位点原子的电子态密度,结果如图4所示。可见,未掺杂和掺杂Ga或Sn的Al2O3表面Al原子的3p轨道和Cl原子的3p轨道均有重合杂化现象,表明Cl与Al(1)-1位点的Al原子之间形成了化学键,这与其他研究结果[19]一致。

图4 未掺杂和掺杂Ga或Sn的Al2O3表面Al(1)-1位点原子的电子态密度Fig.4 Electronic density of states of atoms at Al(1)-1 site on the none-doped and Gaor Sn-doped Al2O3surfaces
一般认为,费米能级处的电子较为活跃,该位置态密度越小,电子结构越稳定。由图5中Al原子的3p轨道电子态密度可以看出,相较于Nonedoped表面,Ga-doped表面Al原子的3p轨道下移,远离费米面,电子能量更低,因此和Cl的电子耦合后释放的能量更少,吸附能升高;Sn使表面Al原子的3p轨道上移,靠近费米面,电子能量更高,因此和Cl的电子耦合后释放的能量更多,吸附能下降。由表1可知,Cl在含Ga的Al2O3(0001)表面Al(1)-1位点的吸附能高于Nonedoped表面,在含Sn的Al2O3(0001)表面Al(1)-1位点的吸附能则低于None-doped表面,这与电子结构的分析结果一致。对于其他吸附位点,由于受表面结构的影响,其吸附能与表面原子的电子结构性质对应不明显,但这些位点的吸附同样也受到原子能带重新分布的影响。因此,Al2O3表面掺杂元素后,表面Al原子p能带结构发生变化,导致表面对Cl的吸附能力发生变化。
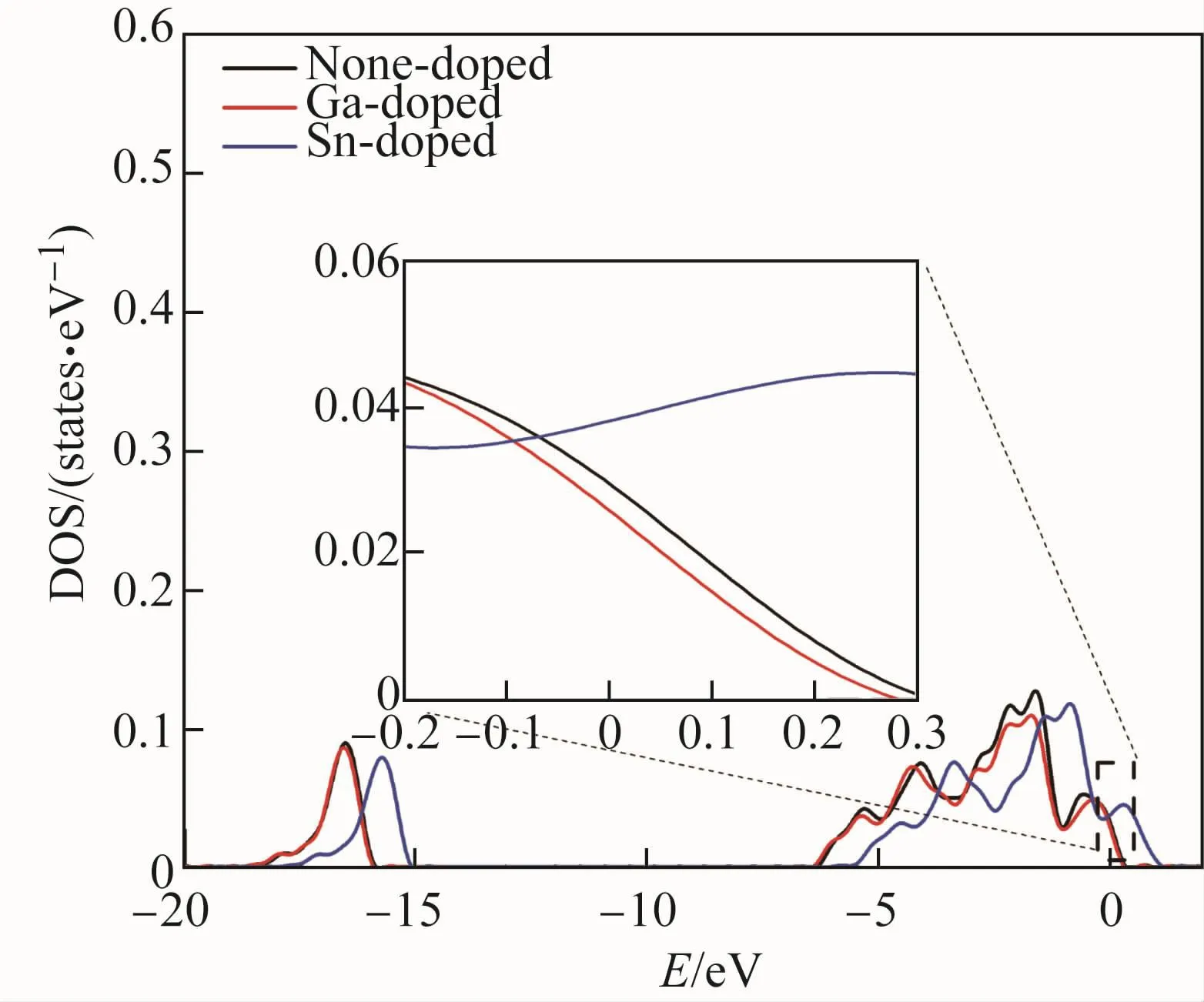
图5 未掺杂和掺杂Ga或Sn的Al2O3表面Al原子3p轨道电子态密度Fig.5 Electronic density of states of 3p orbital of Al atoms on the none-doped and Ga-or Sn-doped Al2O3surfaces
2.3 Ga和Sn对Al2O3(0001)表面耐Cl-腐蚀性能的影响
表2为水环境中不同类型掺杂表面Cl原子最稳定吸附结构的最外层Al原子与第二层O原子之间的键长和原子层间距。可以看出,与None-doped表面Al—O键相比,Ga-doped表面Al—O键长基本不变,层间距仅增加了0.009 1 Å;Sn-doped表面Al—O键长略有增加,层间距也增加了0.154 0 Å。这表明Ga掺杂对Al2O3表面Al—O键强度以及最外层Al原子层与基体之间的结合强度影响不大,而Sn掺杂削弱了表面Al—O键,降低了最外层Al原子层与基体之间的结合强度。
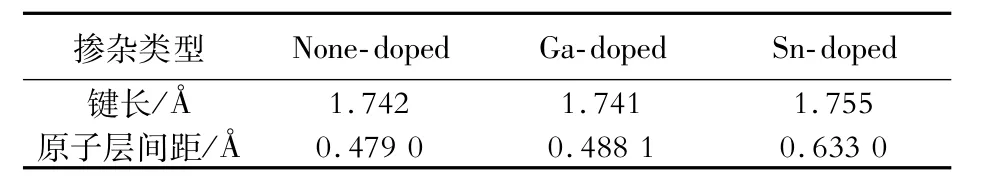
表2 未掺杂和掺杂Ga或Sn的Al2O3表面Al—O键长及原子层间距Table 2 Bond length and atomic layer spacing of Al—O on the none-doped and Ga-or Sn-doped Al2O3surfaces
因此,Ga掺杂虽然没有改变Cl原子在Al2O3表面的最稳定吸附状态,但使Cl原子吸附能升高,且对表面Al原子与O原子之间的结合强度影响不大。总之,Ga掺杂降低了Al2O3表面对Cl原子的吸附能力,使Cl原子需要更大的驱动力才能与表面原子间发生吸附作用并成键。Sn掺杂Al2O3表面后,不仅降低了Cl原子在Al2O3表面的吸附能,使表面更容易吸附Cl,还明显降低了Al2O3表面Al原子与O原子以及基体之间的结合强度,使Al的解离倾向增大,这可能是Sn降低铝溶解点位的微观原因[20]。
3 结论
掺杂Ga或Sn的Al2O3表面Cl最稳定的吸附状态均为化学吸附,Ga提高了Al2O3表面Cl原子的吸附能,且不明显改变Al2O3的表面结构,有助于提高铝合金的耐Cl-腐蚀性能。而Sn不仅降低了Al2O3表面Cl原子的吸附能,还降低了表面Al原子与基体之间的结合强度,导致Al2O3的耐Cl-腐蚀性能下降。Al2O3表面掺杂合金元素后原子能带的重新分布是导致其对Cl原子吸附性质不同的主要原因。
致谢:感谢上海大学高效能计算中心,上海智能计算系统工程技术研究中心(No.19DZ2252600)提供的计算资源和技术支持。
猜你喜欢
山东冶金(2022年4期)2022-09-14
山东冶金(2022年4期)2022-09-14
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化·中考版(2021年10期)2021-11-22
农业环境科学学报(2021年4期)2021-05-25
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
中国金属通报(2020年1期)2020-04-23
新高考·高一物理(2015年6期)2015-09-28
