
药物免修饰的药靶鉴定蛋白质组方法研究进展
2021-09-23 09:26吕佳纹叶明亮
质谱学报 2021年5期
吕佳纹,叶明亮
(1.中国科学院大连化学物理研究所,辽宁 大连 116023;2.中国科学院大学,北京 100049)
现代药物研发领域最常用的2种新药筛选策略是表型筛选和靶向筛选[1-2]。表型筛选是一种直接通过表型与疾病的联系来评价药物疗效的直观方法,曾在上世纪众多经典药物的发现中起到巨大作用。2000年左右,研究者们使用靶向方法筛选到了许多有治疗价值的明星分子,因此该方法在药物发展领域得到了更多重视。靶向筛选方法一般是从已知的“成药”靶蛋白出发,筛选一系列候选小分子化合物或其他抗体药的药物文库,从而寻找对成药靶蛋白有调控作用的治疗潜力的药物分子。在过去10年中,表型筛选在新药研发领域中掀起了一股复兴狂潮[3-4]。尽管21世纪初期是一个靶向筛选占主流的时代,但近十几年来,表型筛选在发现新药小分子化合物方面的贡献大大超过了靶向筛选[5]。然而,表型筛选最大的问题是其获得的候选化合物的蛋白质靶点是未知的[6]。虽然在药物开发的早期阶段并非必须知晓药物的靶蛋白,但是若要充分研究候选药物的药效及其原理,构建药物靶点蛋白和脱靶蛋白数据库仍然是非常关键的[7]。通过深入研究药用小分子化合物在细胞内的作用机制(mechanism of action, MoA),建立“药物结构-作用靶点-表型”的三元模型,有助于更深入透彻地理解药物分子是如何参与调控生物体内的生命活动[8]。尤其是药物分子与其靶蛋白之间相互作用的研究,即靶点解析(target deconvolution),在临床用药指导、患者分级策略、不良反应预测以及推广临床使用等临床应用领域具有重大意义[9]。
在很长一段时间内,基于遗传学的配体-蛋白相互作用解析方法被广泛应用于识别新的药物靶点和研究在药物功能和耐药性中起作用的基因[10]。然而,这些基于遗传学的方法只能间接地为药物-靶蛋白相互作用提供证据。遗传学的研究方法通常涉及抑制或过表达相关基因来研究药物小分子对生物体表型的影响,因此,通常是假说驱动的验证性研究。除此之外,基于基因组学和转录组学的研究方法都缺乏诸如翻译后修饰(PTM)、蛋白质定位和蛋白质-蛋白质相互作用等信息。相反,在蛋白质组学数据中,这些信息得以保留。随着蛋白质组学的重要分析工具——质谱仪的发展,尤其是Orbitrap系列质谱仪分析能力的提升,极大拓展了蛋白质组的鉴定深度和定量精度。在过去的15年中,基于质谱的蛋白质组学和基于计算技术的生物信息学领域的进步,极大地推动了药靶筛选的研究,发展了一系列基于蛋白质组学的靶点解析方法。
在化学蛋白质组学方法中,亲和色谱法(affinity chromatography)是一种经典的策略,被广泛应用于研究蛋白质与生物活性小分子的相互作用或蛋白质-蛋白质的相互作用[11]。该方法通过适当的官能团(通常是羧酸或伯胺),将配体分子固载于固相基质上,然后与蛋白质提取物共孵育,经过几个洗涤步骤后,与配体结合的蛋白保留在基质上,通过蛋白变性或自由配体竞争将这些结合蛋白洗脱下来,最后使用凝胶电泳或液-质联用技术对洗脱的蛋白质进行分析[7]。这种方法的一个明显缺点是需要合成待研究活性化合物的衍生物,并将其以化学偶联的方式固载到基质树脂材料上。这一化学衍生过程可能会影响待研究分子的生物活性部分。此外,被固载的配体分子的蛋白质结合特性(即特异性和/或亲和力)可能与游离配体有差异,因此无法将目标蛋白质捕获到树脂上。另一个问题是,因为引入了连接臂和固载树脂材料,可能会有许多非特异性蛋白被吸附在固相基质上,对后续的样品分析造成一定的干扰[12]。还有一种专门研究蛋白激酶抑制剂的化学蛋白质组方法,与亲和色谱法在策略上非常相似。即先将一系列泛激酶抑制剂分子通过共价键固载到基质材料上,再将待研究的抑制剂小分子和固载基质材料共同投入到样品中与蛋白共孵育,使待研究的小分子与泛特异性亲和性基质共同竞争结合蛋白,从而检测待研究的抑制剂与其靶蛋白的结合能力[13-14]。这种方法具有广谱性,能够实现超过200个蛋白激酶靶点的检测,目前泛激酶抑制剂固载基质已被成功商品化。这种方法不需要对配体小分子进行化学衍生,而是通过检测待研究的抑制剂小分子与固载的泛激酶抑制剂之间对靶蛋白竞争能力的差别,反向地推断配体分子与靶蛋白的结合情况[15]。这种基于Kinobeads方法的实施过程相对比较复杂,其实验过程需要使用昂贵的标记试剂进行定量检测[12]。
另一种广泛使用的方法是由Cravatt团队开发的基于活性的蛋白质组分析(ABPP),利用化学探针检测与某一类配体小分子有亲和作用的蛋白质的化学蛋白质组[16-18]。ABPP方法最初用于酶谱分析,后来被引入药物靶点筛选领域。这种方法利用了特殊设计的化学探针,其结构通常包括3个功能部分:1) 结合基团。与靶蛋白有亲和相互作用,将探针引导至受体结合区域;2) 反应基团。使探针与受体蛋白质发生共价连接;3) 报告基团。通常为可拓展的基团,如叠氮/炔基等,或是荧光基团、可富集基团等可被检测的标签,用于定性、定量地检测结合蛋白[16,19]。常规的分析流程为:通过反应基团将探针与靶蛋白共价连接后,使用点击化学技术将报告基团接到探针上,原位发光或质谱等方式检测探针捕获的结合蛋白。这些亲和性探针能够以化学键的方式“冻结”小分子与靶蛋白之间的相互作用,可以帮助固定一些低亲和力的靶蛋白,提高对这些相对较弱结合力靶点的鉴定能力[7]。
这些化学蛋白质组学技术是高通量药物靶点筛选的有力工具[3,20-22]。然而,值得注意的是,这2种方法都需要额外对小分子药物或竞争性抑制剂进行化学衍生,随之带来了一系列问题。例如,一些小分子化合物在化学结构上缺乏合适的化学修饰空间,因此无法被设计衍生成为化学探针或固载有药物小分子的固相基质[23]。额外的化学衍生还可能影响药物的生物活性/靶点结合特异性,造成靶蛋白的错误识别或无法检测到与小分子原有结构发生结合的蛋白[24-25]。基于亲和色谱法利用了固定化小分子药物与靶蛋白之间的亲和作用来捕获靶蛋白,因此,在检测低丰度蛋白和亲和力较弱的靶蛋白上存在着一定难度。而ABPP技术利用化学探针直接标记、富集和鉴定靶蛋白,在一定程度上缓解了这一问题。由于探针连接到某些蛋白质氨基酸残基上时,可能会影响胰蛋白酶的消化效率,酶解产物肽段可能过长而不适合bottom-up蛋白质组学分析。此外,有一些探针化学结构不稳定或空间位阻过大,导致质谱分析时探针标记的肽段无法被有效地碎裂,造成质谱分析困难。因此,有必要发展无需对药物分子进行化学改造的药靶筛选方法。
在过去的几十年中,蛋白质组学家联合生物学家发展了一系列药物免修饰(modification-free)的药物靶蛋白分析方法[26],这些方法不需合成探针或对药物分子进行任何化学修饰。相反,它们可以通过直接测量药物结合后蛋白质性质的特定变化鉴定药物靶蛋白,如测量蛋白的热稳定性和蛋白酶解敏感性变化。这些蛋白质性质的改变是由于药物结合后,靶蛋白能量状态、蛋白折叠结构等生物物理性质发生了改变。使用诸如细胞热位移分析、热蛋白质组分析、蛋白质氧化速率稳定性分析、基于有机溶剂沉淀蛋白稳定性方法、药物亲和相应靶点稳定性分析、脉冲酶解法、限制性酶解法等药物免修饰的方法,即可区分与药物分子发生结合的靶蛋白和游离蛋白。本文将详细阐述以上方法的原理、实施和应用。
1 药物免修饰的药物靶蛋白筛选方法
1.1 细胞热位移分析(热蛋白质组分析)
热位移分析(TSA)是使用最广泛的非化学修饰药物靶点的发现方法之一[27]。通过测量光散射[28]或荧光[29]的变化来确定蛋白熔融点的变化,并绘制药物结合蛋白和游离蛋白的热熔融曲线。当靶蛋白与药物发生结合形成复合物时,靶蛋白的热稳定性增强,通过比较游离态和结合态的靶蛋白热熔化曲线中点的位移(Tm),即可判断蛋白是否为药物的靶点[30]。该方法简单明了,但由于探针不能区分不同的蛋白质,因此,TSA仅适用于纯化蛋白的分析。为解决这一问题,Nordlund团队开发了一种概念类似的技术,称为细胞热位移分析(CETSA),用于在活细胞中直接研究药物与靶蛋白的相互作用[31]。在CETSA方法中,用药物或载体处理多份细胞裂解物或完整细胞,加热至10个不同温度点并冷却,然后高速离心分离含有可溶性蛋白的上清液部分和不溶性的沉淀部分,示于图1。随着温度升高,蛋白质逐渐展露出疏水核,使蛋白质在高温下沉淀。因此,可通过测定随温度升高的可溶性蛋白质组分来衡量蛋白质的稳定性。在CETSA中,不同温度点可溶性组分中的蛋白质分别用蛋白免疫印记法定量检测,然后根据蛋白质的强度随温度升高的变化,绘制蛋白质的热熔化曲线。根据药物结合蛋白和游离蛋白之间的热熔化曲线中点位移区分药物靶蛋白[32]。在确定蛋白热熔化中点位移温度后,可通过等温剂量依赖的实验研究药物剂量与药物靶蛋白结合之间的关系,从而测定药物分子与靶蛋白相互作用的亲和力[31]。在这种等温剂量响应指纹图谱方法(ITDRFCETSA)中,细胞或细胞裂解产物暴露于不同浓度的药物处理条件下,同时保持加热时间和温度恒定,通过比较蛋白结构受药物保护的程度,可以识别出该蛋白是否为真正的药物靶点,并且推断对应的Kd值。CETSA技术具有操作简便、与完整细胞相容性好等优点,在全球的学术界和工业界都已得到广泛应用,是目前药物靶蛋白筛选的常用方法之一[33-36]。此外,CETSA还被应用于研究脱靶蛋白、结合机制、药效和细胞耐药性等生物学问题[2,37]。除了能定性鉴定药物-靶点的相互作用外,ITDRFCETSA还可用于探索动物体内剂量依赖的靶点相互作用、帮助确定药物的使用和剂量、监测耐药性[31]。
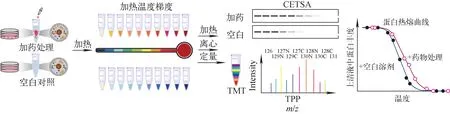
图1 细胞热位移(热蛋白质组分析)方法工作原理及流程Fig.1 Principle and workflow of CETSA (or TPP)
最初,CETSA技术使用基于抗体的蛋白质免疫印记法(western blotting)或近程放大发光均相检测(alpha screening)进行蛋白定量分析,从而描绘蛋白在加药与否条件下的热融化曲线。这种基于抗体定量的CETSA方法需要提前了解目标靶蛋白并获得对应抗体,因此不适用于无偏好性地(unbiased)发掘药物新靶点,也无法获得蛋白质组水平的全局化数据。2014年,Savitski团队[38]将CETSA方法与多重定量质谱法相结合,发展了热蛋白质组分析(TPP)方法,并应用于药物靶点蛋白和脱靶蛋白的鉴定。利用基于串联质量标签(TMT-10plex)标记的高通量鉴定和定量能力,TPP方法能够同时检测全蛋白质组在10个温度下的热变性状态,并绘制完整的熔化曲线,从而可以确定每种蛋白质的热位移变化。本质上,TPP与CETSA的原理完全一致,都是基于蛋白质受热变性会逐渐展开结构并析出沉淀;不同之处在于,TPP使用多重标记的质谱技术进行蛋白定量,最初的CETSA使用基于抗体的定量技术。因此,这种基于高通量质谱进行蛋白定量的热位移技术,又被称为MS-CETSA。与CETSA相同,TPP/MS-CETSA可被用于细胞提取物层次的研究,亦可被应用于完整的活细胞层次,通过结合这两方面的信息,可以区分由于靶蛋白直接与配体结合引起的热位移和下游信号通路变化引起的蛋白质热位移[39]。那些在活细胞实验中热稳定性受到影响、而在细胞提取物中未受影响的蛋白质可能是药物分子的间接靶点[40]。因为质谱方法的分析通量很高,可以一次性分析成千上万的蛋白,TPP方法广泛应用于筛选一系列潜力药物或其他小分子的靶蛋白及其相互作用蛋白与下游信号通路的研究[41-45]。还有一种方法——配体稳定靶点识别技术(TILS)[46],与CETSA或TPP方法研究可溶性部分蛋白的不同,该方法研究的是蛋白在加热之后产生不可溶的沉淀部分,可被视为CETSA/TPP的互补方法。
虽然CETSA/TPP方法最初是为研究蛋白与药物相互作用而开发的,这2种基于热稳定性变化的方法也可被用于研究蛋白与其他配体分析的相互作用,例如蛋白-蛋白的相互作用,蛋白与核酸、代谢物、氨基酸、脂质等生命活动中常见配体分子的相互作用。2018年,开发MS-CETSA和TPP的2个团队分别将这种基于热稳定性变化的方法应用于细胞周期蛋白质组的研究,通过检测细胞生命周期过程中蛋白质丰度和热稳定性的变化,揭示了在细胞生命周期不同状态下全蛋白质组及蛋白-蛋白相互作用网络的变化。这种针对“不同细胞状态×不同加热状态”两个维度下的热稳定性变化研究,被称为2D-CETSA或2D-TPP,即“温度梯度(temperature)×处理状态(condition)”的二维样品阵列。Pär Nordlund团队将这种策略应用到细胞周期不同状态下蛋白质与其他配体的相互作用研究中,如蛋白质-蛋白质的相互作用,蛋白质与核酸、代谢物、脂质、辅因子等配体的相互作用[47]。该研究首次将基于热稳定性的蛋白质能量状态变化引入细胞周期蛋白质组,探究细胞生命周期中不同时期的蛋白质热稳定性和表达水平变化,并将发生动态变化的蛋白分为3类:1) 仅在蛋白表达水平发生变化;2) 仅在蛋白热稳定性水平发生变化;3) 在蛋白表达和热稳定性水平都发生变化。研究结果表明,发生了热稳定性变化的蛋白相比仅发生表达水平变化的蛋白参与了更多的蛋白质相互作用网络。除蛋白表达水平外,CETSA技术能够通过测定热稳定变化,从而反映细胞生命周期不同状态下蛋白质实现多种生物学功能的过程,有助于研究细胞周期过程中蛋白质相互作用的动态变化。研究人员还指出,处于同一相互作用网络中的蛋白质有相似的热稳定性位移,表明这些蛋白可能作为1个蛋白质复合物执行特定的生物学功能。在后续工作中,该团队开发了热稳定性近程共聚集(TPCA)算法,并用于研究已知蛋白复合物,证实了这些已知的蛋白复合物确实能表现出“热稳定共聚集”效应,为系统性地预测蛋白质复合物以及研究细胞异质性的蛋白复合物提供了很好的工具[48]。Savitski团队[49]也将这种2D-TPP策略应用于系统性地研究细胞周期过程中蛋白质的热稳定性变化和可溶性蛋白丰度变化,证明了在细胞周期的M期和G1期之间,细胞中蛋白的热稳定性变化是广泛存在的。他们认为细胞生命周期过程中,蛋白丰度变化和蛋白热稳定性变化是相互独立的,换言之,细胞周期相关的蛋白热稳定性变化是通过与蛋白丰度无关的机制调控的。
然而,这些基于热稳定性变化的方法存在一定的局限性。首先,由于需要保持蛋白质活性,在蛋白提取过程中通常不添加变性试剂,使得膜蛋白等高疏水性蛋白质无法检测。实际上,很多药物的作用靶点均存在于细胞膜上。为解决这一问题,可以向细胞提取液中添加温和的洗涤剂(如NP-40)来帮助溶解膜蛋白,在一定程度上提升膜蛋白的溶解性[50]。此外,这些方法不适用于解折叠后仍不发生沉淀聚集的蛋白[51]。理论上,蛋白质的热稳定性在一定程度上会受到配体结合的影响。然而,一些蛋白质在表观熔化温度上并没有明显受到配体结合的影响[39]。对于一些多结构域蛋白或结构巨大的蛋白,配体诱导的结构变化可能只发生在很小的范围内,不足以对蛋白质聚集和沉淀产生影响[32,39]。除此之外,观察到一些蛋白质在药物处理后热稳定性降低,发生了去稳定化作用[52]。一种解释是这些蛋白质并不是药物直接的靶蛋白,而是真正靶蛋白的相互作用蛋白。在实施过程中,CETSA或TPP方法的单个样品分析通常需要10个温度点,如果考虑到不同处理状态或来源的样品与生物学/技术重复,通常需要10×N个样品测量,过大的分析通量对于基于质谱的蛋白质组分析的稳健性是一个严峻的挑战。最近,研究者们分别从样品前处理和数据处理两个方面出发,发展了一系列优化热稳定性分析的方法,旨在降低待分析样品数目。一种近年来比较流行的“一锅法”策略是将多个加热温度点的样品混合后直接分析单个合并样品,将CETSA原有的测定热融曲线的“位移变化”转变为“积分变化”[53]。本课题组开发了一种微球辅助的热致沉淀分析方法(MAPS),在样品前处理过程中,引入微球材料捕获蛋白质受热沉淀,随后进行基于微球的快速、高通量的SP3法样品前处理;从热稳定性数据处理方面,开发了一种不依赖于拟合热熔曲线的数据处理策略,通过比较加药/空白两种处理条件之间的整体稳定性差异,在数据处理层次上“合并”多个温度点的数据,降低所需分析样品数目的同时,放大了热稳定性变化差异[54]。另一种等温热位移(iTSA)方法[55]为在单一温度点下进行热稳定性实验,通过增加样品重复次数提高靶蛋白鉴定的统计学可信度,在单一温度下,也可达到与CETSA/TPP相似的效果,但该方法不能大幅度减少所需分析的样品个数。
1.2 蛋白质氧化速率稳定性分析
上述基于热稳定性方法反馈的是蛋白水平的信息,仅能检测到蛋白与配体结合后的整体生物物理性质的变化,无法揭示配体结合域的详细信息。Fitzgerald团队专注于蛋白质结构及其功能的研究,在2000年发展了一种基于氢氘交换(H/D交换)速率方法,用于全局性检测蛋白质上的酰胺基团,以分析蛋白质稳定性[56]。因为某些基团上的不稳定氢原子可以与周围溶剂中的质子发生自由交换,通过监测这些氢原子与重水溶剂中氘原子的交换速率来考察蛋白质的稳定性。然而,H/D交换过程中的回交现象会对蛋白结构折叠的观测产生不利影响,随后该团队发展了一种与先前的酰胺H/D交换策略相似的方法,即基于蛋白质氧化速率稳定性(SPROX)的配体-蛋白相互作用研究方法[57]。在SPROX中,蛋白质的稳定性是通过甲硫氨酸残基的氧化速率与化学变性剂(如盐酸胍或尿素)浓度的关系来评估的。向含有或不含药物的细胞蛋白质提取物中加入一系列浓度递增的化学变性剂(例如盐酸、GdmCl)缓冲液,使用等量的过氧化氢试剂氧化蛋白样品后,对蛋白质氧化反应进行猝灭,再进行常规bottom-up蛋白质组样品前处理,随后进行质谱定量分析,测定未氧化和氧化的含甲硫氨酸肽段的相对强度,并绘制含甲硫氨酸肽段氧化程度随SPROX缓冲变性剂浓度变化的曲线。通过加药或空白处理下的含蛋氨酸的氧化肽和非氧化肽的氧化曲线的中点位移,可以判断该肽段所归属的蛋白是否为药物直接或间接相互作用的蛋白,示于图2。曲线中点位移可用于计算蛋白质折叠自由能(ΔGf)、结合自由能(ΔΔGf)和解离常数(Kd)[57-58]。原则上,SUPREX和SPROX均测量蛋白质的去折叠/复性平衡。与H/D交换的可逆问题相比,SPROX的主要优点是化学反应的不可逆性。SPROX方法开发早期,由于受到定量通量的限制,最初主要针对单个纯化蛋白质进行蛋白折叠情况研究[57],但随着基于质谱的蛋白质组学技术的飞速发展,该方法被拓展应用于研究复杂生物样品中的蛋白质-配体结合特性[59]。

图2 蛋白质氧化速率稳定性分析方法工作原理及流程Fig.2 Principle and workflow of SPROX
理论上,任何定量方法都可以在SPROX中定量分析非氧化和氧化含蛋氨酸的肽段。然而,考虑到SPROX中的多重变性条件,基于稳定同位素标记的多重定量蛋白质组学技术更适合作为SPROX的定量手段,包括串联质谱标记(TMT/iTRAQ)的化学标记[60]和细胞培养中氨基酸的稳定同位素标记(SILAC)[61-65]。2019年,有研究[62]使用SPROX方法与TMT联用,将其命名为高分辨SPROX(HR-SPROX),报道了在人类细胞提取物中检测到的约3 000个蛋白质中约10 000个独特的折叠稳定性变化区域。2020年,Fitzgerald团队[66]将SPROX技术与基于热稳定性的CETSA方法相结合,用于研究抗组胺药物氯马斯汀的靶蛋白和药效机制,成功鉴定并验证了疟原虫伴侣蛋白TRiC/CCT是氯马斯汀的靶点。与CETSA/TPP方法相同,SPROX方法也需要一系列不同浓度变性剂处理的样品点来描绘蛋白/肽段的氧化曲线,对后续的蛋白质组质谱分析通量造成很大压力。“一锅法”的思想可被应用于所有基于多测量点拟合蛋白稳定性曲线的非化学修饰的药物靶蛋白筛选方法,同样地,也可被应用于SPROX分析中,将多个变性剂浓度处理后的样品合并,“多点分析”转化为“单点分析”,可以降低样品数目、提高分析通量[67]。
SPROX方法最初的设计是用于研究蛋白质与药物的亲和力,现在被扩展到蛋白质与其他小分子配体的相互作用[60,68]、疾病相关的蛋白质结构变化和翻译后修饰[61,69]、金属离子与蛋白的相互作用及毒性机制[70]等研究。SPROX的局限性之一是其只适用于研究含有甲硫氨酸残基的蛋白质。为克服这一弱点,开发了一系列针对其他氨基酸残基的化学修饰方法,包括针对赖氨酸残基的SMTA[71]、针对色氨酸的HNSB[72]。与CETSA和其他基于热稳定性的方法相同,SPROX主要适用于检测药物与可溶性蛋白质的相互作用。由于该方法需要使用外源的化学变性剂和氧化剂,无法像CETSA/TPP一样被应用于分析活细胞。SPROX可在肽水平上分析氧化和非氧化蛋氨酸,表明其具有揭示每个独特蛋白质结构域的局部稳定性的能力。但由于肽段水平定量的信噪比、假阳性等问题,这种针对局部结构域的方法在药物靶蛋白的鉴定覆盖度和准确性上有所逊色。因此,Fitzgerald团队开发了一种化学变性相关的蛋白沉淀方法(CPP),利用化学变性剂使蛋白沉淀,随后稀释变性剂浓度使已被展开结构的变性蛋白析出沉淀,通过检测药物对其靶蛋白结构的保护效应,实现高覆盖的药物靶蛋白鉴定和筛选[73]。
1.3 基于有机溶剂沉淀蛋白稳定性方法
目前,已开发了许多利用蛋白质沉淀过程解析药物靶蛋白的方法。如CETSA/TPP、SPROX等都是在蛋白变性过程中,检测药物结合对蛋白质稳定性的影响。此前,本课题组发展了一种基于有机溶剂沉淀蛋白稳定性方法(SIP)[74],在蛋白质组层面进行药物靶蛋白筛选,示于图3。该方法使用蛋白沉淀中常用的丙酮沉淀剂溶液(50%丙酮-50%乙醇-0.1%乙酸),使蛋白在一系列有机溶剂浓度(9%~19%)中逐渐沉淀,因与药物发生结合的靶蛋白会比游离蛋白更稳定,更不易被有机溶剂沉淀,通过检测上清液中剩余蛋白质的相对强度,即可揭示全蛋白质组在受到有机溶剂影响下的蛋白稳定性变化。使用SIP方法对格尔德霉素的潜在靶蛋白进行筛选,不仅鉴定到已知靶蛋白HS90AA1、HSP90AB1和HSP90B1,还筛选出若干个潜在的脱靶蛋白。在这些候选靶蛋白中,用蛋白印迹法(WB)验证了筛选到的NDUFV1确实是格尔德霉素的脱靶蛋白。与基于热稳定性的CETSA和基于氧化的SPROX方法相同,SIP方法也能测量药物与其靶蛋白的亲和力。用该方法评估了格尔德霉素与其已知靶蛋白HSP90AB1的解离常数,不同有机物浓度、不同剂量格尔德霉素浓度下的实验结果表明,格尔德霉素与HSP90AB1的半饱和浓度在0.1~1 μmol/L之间,与前人报道的Kd=1.2 μmol/L相吻合。我们也对筛选的候选靶蛋白NDUFV1的Kd进行了评估,发现半饱和浓度约为10 μmol/L,其与格尔德霉素的亲和力比与已知靶蛋白HSP90AB1的弱。

图3 基于有机溶剂沉淀蛋白稳定性方法工作原理及流程Fig.3 Principle and workflow of SIP
还有一种DiffPOP方法,使用甲醇/乙酸沉淀剂促进蛋白沉淀,鉴定到丝氨酸羟甲基转移酶SHMT2是组蛋白去甲基转移酶抑制剂小分子JIB-04的作用靶点[75]。这些基于有机溶剂沉淀蛋白的方法均无法直接在活细胞层次进行研究,而需要将细胞裂解后提取蛋白进行体外实验。因蛋白变性的机理不同,基于有机溶剂沉淀稳定性的SIP方法与基于热稳定性的CETSA方法具有很好的互补性。
1.4 药物亲和响应靶点稳定性分析
与药物发生结合的蛋白比游离蛋白更不易被蛋白酶水解[76-77]。从热力学角度,因与药物发生结合后蛋白的能量状态降低,药物-蛋白复合物从折叠态到非折叠态所需跨越的能量势垒更大,因此,药物结合蛋白的能量状态更稳定。从动力学角度,这些能量状态更稳定的药物结合靶蛋白的解折叠速率比游离靶蛋白低,暴露可酶切位点的可能性更低,因此,药物结合蛋白上的酶切位点更不容易被蛋白酶所触及。而蛋白质的水解敏感性取决于这些可酶切位点的可接触范围,同样条件下药物处理后的靶蛋白比未经药物处理的靶蛋白更不易被蛋白水解酶降解。早在上世纪90年代就已经提出使用限制性酶切方法研究蛋白的折叠速率[78-79],在2009年,Huang等[80-81]利用药物结合与否会影响蛋白折叠-解折叠速率这一特点,开发了药物亲和反应靶点稳定性(DARTS)方法用于药物靶点筛选。DARTS方法中,用目标药物或空白溶剂处理蛋白质样品,然后用蛋白酶对细胞裂解物中的蛋白质进行限制性酶解,最后用SDS-PAGE对样品进行分离,示于图4。与溶剂对照组相比,被小分子结合保护的蛋白更不易被蛋白酶水解,因此,其蛋白条带强度比对照组高。DARTS法可以分析纯化蛋白或全细胞裂解后的蛋白提取物,经SDS-PAGE分离后,可根据不同目的采用蛋白印迹法或质谱法定量分析样品组和对照组之间的蛋白定量变化[80,82]。

图4 药物亲和响应靶点稳定性分析方法工作原理及流程Fig.4 Principle and workflow for DARTS
1.5 脉冲酶解法
在DARTS方法中,蛋白质通常在天然条件下进行限制性酶解。然而,有些蛋白质即使经过几天的消化也能保持紧密结构[83],可能是因为这些蛋白质在天然条件下紧密折叠,蛋白酶无法接触到特异性酶切位点,此时,DARTS无法识别这些蛋白质是否为药物靶点。脉冲蛋白水解法(PP)通过引入变性剂可在一定程度上解决上述问题[84]。与DARTS不同,脉冲蛋白水解法中的蛋白质酶切是在一系列不同变性剂浓度下进行的,蛋白质受到变性剂的影响会发生不同程度的变性和解折叠,示于图5。在这种方式下,即使是结构最紧密的蛋白质也可以在高变性剂浓度(如8 mol/L尿素)下完全展开;然后,将蛋白酶添加到上述溶液中进行脉冲水解,即短时间消化酶切已经发生解折叠的蛋白质,但保持折叠结构的蛋白质不易被蛋白酶接触到特异性酶切位点,抵抗蛋白酶解[84]。在SDS-PAGE分离得到的样品中,未被酶切的完整蛋白质可以通过染色法进行定量。因此,可以确定保留折叠结构的蛋白质,即剩余的蛋白质部分,并且可以绘制该部分与变性剂浓度的相关曲线。同SPROX一样,PP法引入了离液剂使蛋白变性,蛋白变性曲线是在离液剂浓度递增的条件下描绘的,可用于计算蛋白的全局稳定性,即蛋白折叠自由能。

图5 脉冲酶解法工作原理及流程Fig.5 Principle and workflow of PP
在DARTS和PP方法中,SDS-PAGE经常被用于蛋白的分离和定量检测,随后切下发生强度变化的蛋白泳道胶条进行后续的MS分析。然而,一些生物样品的复杂性过高,许多蛋白质的分子质量相近且聚集在一起,目标蛋白可能被高丰度的非目标蛋白掩盖,用肉眼很难找到差异变化的蛋白泳道[85]。为了克服这一困难,更高分辨率的二维凝胶电泳被应用于脉冲蛋白水解方法[86]。除了通过凝胶条带强度测定蛋白丰度外,还可使用基于LC-MS/MS的定量蛋白质组学平台,使用串联质量标签[87]、细胞培养中氨基酸的稳定同位素标记(SILAC)[88]等技术定量每个蛋白质样品中完整蛋白质的相对丰度。DARTS和脉冲蛋白水解法的原理都是基于检测配体诱导的蛋白稳定性变化。在无变性剂(DARTS)或有变性剂(脉冲蛋白水解)的情况下,药物结合可以保护靶蛋白不被蛋白酶消化。由于这2种方法测定的都是酶解剩余的完整蛋白质,而不是酶解产物肽段,因此,不能用于揭示药物与靶蛋白的结合位点。并且这2种方法都需要使用外源的蛋白水解酶,所以都无法应用于活细胞的分析。
1.6 限制性酶解法
限制性酶解是一项经典的生物技术手段,DARTS法和脉冲水解法本质上都利用了限制性酶解技术。2004年,Fontana等[79]提出了一个“天然结构-解折叠结构”平衡理论来解释限制性蛋白水解过程。在该理论中,蛋白质的部分未折叠区域(例如柔性环)具有更高的部分结构可变性,从而为蛋白酶活性位点的结合提供了更多的可接近性[79,89]。限制性酶解法用于蛋白质结构的研究由来已久,但因为缺乏有效的分离检测手段,在很长一段时间内该方法都被局限于纯化的单一蛋白的研究[90-93],尤其专注于酶解反应后剩余的完整蛋白,而非酶解产物。上述DARTS法和脉冲水解法研究的是经短时间酶切后样品中剩余的蛋白质,仅能提供蛋白层面的信息,而酶切位点相关的结构域信息被忽视了。2014年,在前人研究的基础上,Picotti团队创造性地提出了一种基于两步消化的限制性酶解方法,并结合基于质谱的定量蛋白质组技术研究蛋白质的结构,被称为限制性酶解技术(LiP)[94],示于图6。使用这种方法在非变性条件下提取到的天然蛋白质需经过两步酶切过程:1) 限制性酶解,使用非特异性蛋白酶(例如,蛋白酶K、嗜热菌蛋白酶等)在较低浓度和较短时间内进行限制性蛋白水解,经过水解产生大的蛋白质碎片;2) 经过上一步限制性水解得到的大块蛋白碎片被转移到变性条件下,以猝灭第一步的蛋白质水解,经胰蛋白酶充分消化以产生适于bottom-up蛋白质组学分析长度的肽段。此外,还需1对外参样品,即与样品组和对照组来源完全相同的1对样品直接进行经典的定量蛋白质组分析,蛋白样品经变性、烷基化、胰蛋白酶消化后送入质谱分析,这对样品将作为外源参照标准用于样品组和对照组之间蛋白定量的归一化。通过高灵敏度的选择性反应监测(SRM)技术精确地定量样品组和对照组之间肽段相对强度的差异,从而推断蛋白结构差异[94]。该方法关注了限制性水解产生的产物肽段,由于药物结合带来的空间位阻和蛋白解折叠速率变低将导致某些蛋白结构区域无法被蛋白水解酶触及,因此,使加药组与对照组之间的限制性水解产物肽段丰度不同,这些丰度差异的限制性水解位点很可能含有宝贵的药物结合区域信息。

图6 限制性酶解法工作原理及流程Fig.6 Principle and workflow of LiP
随着基于质谱的蛋白质组技术的发展,涌现了越来越多高效的蛋白定量技术,2018年,Paola团队[95]利用经典的鸟枪法蛋白质组label-free和DIA定量技术结合限制性酶解技术,发展了基于限制性酶解的蛋白与小分子互作扫描(LiP-SMap)方法,研究了大肠杆菌和酵母蛋白质组与多种代谢物分子的相互作用关系,首次提出了“含有结构信息的肽段(conformotypic peptide)”概念,揭示了蛋白上与代谢物分子发生结合的区域。2020年,该团队[96]将这种限制性酶切揭示蛋白结构的策略应用到药物靶蛋白筛选以及药物结合区域的研究,使用机器学习算法给出打分值来揭示药物与蛋白结合情况和大致的结合区域范围。2021年,该团队将LiP方法的研究范围拓展到了蛋白翻译后修饰、变构催化、蛋白质-蛋白质相互作用、蛋白质聚集等结构蛋白质组领域,将1个蛋白上含有全部结构信息的肽段列为1个条形码,作为蛋白结构信息的“身份卡”,使用LiP技术即可读取全蛋白质组的蛋白在不同状态下的独特结构信息[97]。值得一提的是,LiP方法还可补充CETSA/TPP技术所缺少的蛋白结构信息,在蛋白加热变性过程中实施限制性酶切,可研究蛋白质的热稳定性、描绘热融曲线以及计算蛋白热熔融点[98]。
综上所述,基于限制性酶切的LiP技术是提供信息最全面的药靶蛋白研究方法,但因其操作步骤多、经两步酶切后产生的肽段样品较复杂等原因,在实际应用中,该方法筛选药物靶蛋白的效果并不如热蛋白质组方法(CETSA/TPP)。我们期待未来蛋白质组样品前处理的简化,随质谱仪器发展的定量准确度和通量的提升,为基于限制性酶解技术的药靶筛选方法带来性能上的飞跃。
2 总结
得益于质谱技术的发展,现代药物靶蛋白已从基于假设驱动的验证性研究发展到蛋白质组水平上无偏好性的高通量靶点筛选。相比于化学蛋白质组学需要在药物分子上进行化学衍生,非化学修饰方法与前沿的定量蛋白质组学相结合,可以在蛋白质组水平上实现高通量、大规模和无偏好性的药物免修饰的靶点筛选。
本文综述了现有的几种基于高通量质谱技术的药物免修饰药物靶蛋白筛选方法,包括基于热稳定性的细胞热位移分析、热蛋白质组分析,基于蛋白解折叠速率的蛋白质氧化速率稳定性分析,基于蛋白酶切稳定性的药物亲和相应靶点稳定性分析、脉冲酶解法、限制性酶解法,和有机溶剂诱导沉淀的稳定性分析。这些方法各有其优势和局限性,根据实际分析需求中的样品类型、检测手段和生物物理性质等差别,可以选择适当的方法,示于图7。在样品类型方面,只有CETSA和TPP既能够直接在活细胞层次进行研究,又可以应用于细胞裂解物研究。其他方法的实验条件不适用于活细胞分析,SPROX、SIP、PP需要外源加入变性剂或沉淀剂使蛋白质去折叠,而DARTS、PP和LiP需要外源添加酶来水解蛋白,这些方法只能在细胞裂解液层次上进行研究。在检测手段方面,本文所列方法都可以采用基于抗体的免疫印记法或基于质谱的定量蛋白质组学方法。最初,基于抗体的定量方法是高通量筛选的重要手段,例如CETSA技术与AlphaScreening的联用极大地提高了筛选通量[33]。随着质谱定量精密度的提高,以及同位素串联质量标签的发展,使质谱定量的通量得到提升,基于质谱的定量蛋白质组技术逐渐成为首选检测手段。

图7 基于蛋白稳定性或酶切的药物免修饰的药靶鉴定方法原理和特性示意图Fig.7 Schematics and characteristics of protein stability-or proteolysis-based modification-free methods
然而,这些非化学修饰的药物靶蛋白筛选方法存在一些缺点。首先,这些方法是在非常复杂的细胞全蛋白质组背景下检测配体药物与蛋白质的相互作用,可能导致鉴定到的蛋白间接受到药物影响。配体药物诱导的靶蛋白改变可能导致蛋白质复合物中其他蛋白的生物物理性质改变,造成混淆。与之相比,ABPP等化学蛋白质组技术可以将探针以共价键的方式连接到蛋白上,直接探测靶蛋白的活性中心,为药物与蛋白质的相互作用提供直接的证据。其次,非特异性蛋白质的干扰也是非化学修饰方法的一个困扰。由于一些低丰度的靶蛋白会被高丰度的非靶蛋白“淹没”,基于鸟枪法的定量蛋白质组学很难检测出低丰度的靶蛋白。此外,这些非化学修饰的方法是通过探测靶蛋白与药物结合后某些物理性质的变化,如热力学稳定性的变化或蛋白结构构象的变化。然而,药物处理并不会使靶蛋白在这些物理性质上都凸显出明显的变化,互补方法的联用将在一定程度上弥补这一缺点。例如,CETSA和LiP的联用可在结构域水平上揭示全蛋白质组的热稳定性变化[97]。
猜你喜欢
中老年保健(2021年3期)2021-12-03
中国生殖健康(2020年7期)2020-12-10
材料科学与工程学报(2016年4期)2017-01-15
中国塑料(2016年7期)2016-04-16
合成化学(2015年4期)2016-01-17
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
合成材料老化与应用(2015年4期)2015-07-25
医学研究杂志(2015年7期)2015-06-22
应用化工(2014年1期)2014-08-16
同位素(2014年2期)2014-04-16
