
超高分辨离子迁移谱技术及应用进展
2021-09-23 09:25徐一仟杨其穆蒋丹丹王卫国李海洋
质谱学报 2021年5期
徐一仟,杨其穆,蒋丹丹,厉 梅,王卫国,陈 创,李海洋
(1.中国科学院大连化学物理研究所,分离分析化学重点实验室,辽宁 大连 116023;2.中国科学院大学,北京 100049)
离子迁移谱(IMS)是一种在电场作用下通过离子在中性气体中迁移,从而实现离子分离与检测的技术[1]。自20世纪30年代首次提出测量离子迁移率的方法后[2],在20世纪60年代中叶,Franklin GNO公司开发出第一台商品化的IMS仪器,并应用于军工化学品的监测[3-4]。此后一直到20世纪70年代,对IMS的理论研究、仪器结构以及化学检测能力的大量开创性研究为现代化的IMS仪器奠定了基础[3,5-6]。自20世纪80年代中叶以来,通过改进样品进样方式、电离方法、离子注入技术以及离子分离技术,大大提高了IMS的分析性能[1]。IMS发展至今已具有三大技术优势[7]:1) IMS可与电离效率较高的大气压化学电离源联用,获得10-12量级的检测限;2) IMS分析可在毫秒量级内完成,且与色谱、质谱分离相正交;3) 离子迁移率K与离子形状、尺寸等结构信息直接相关。基于前2种优势,IMS被广泛用于化学战剂、爆炸物、毒品及危化品的现场快速检测,并发展成为一种主流核心技术[8-11]。如今,在世界各地的机场中,均可见到IMS仪器的身影[12]。配备预分离气相色谱的IMS开始用于复杂样品分析,如食品药品质量控制、工业过程控制、呼出气分析等[13-17]。近年来,得益于后2种技术优势的结合,IMS在生物样品分析中获得了广泛应用[18-20]。一方面,IMS具有毫秒级分析速度,适合与色谱、质谱联用[21],构筑三维甚至四维分离系统[22],满足复杂样品对高选择性检测的需求;另一方面,IMS通过离子迁移率K能够获得离子结构信息,有助于生物分子异构体及不同折叠结构的区分识别[23-24]。
在IMS中,主要依据离子与中性分子发生频繁碰撞所表现出离子迁移率K的差异进行离子分离[1]。离子迁移率K是反映离子自身物理化学属性的综合常数,其不仅与离子的质量m、价态z有关,还与离子碰撞截面ΩD(CCS)有关,示于式(1)。这使得IMS能够在较宽气压范围(约102~105Pa)内工作。
(1)
根据离子分离的实现方式[25],IMS可分为迁移时间离子迁移谱(DTIMS)[26]、行波场离子迁移谱(TWIMS)[27]、阱离子迁移谱(TIMS)[28]、非对称场离子迁移谱(FAIMS)[29]、吸入式离子迁移谱(AIMS)[30]等类型。与色谱类似,IMS的分辨率定义为2个离子峰的分离程度[31]。但在实际应用中,为了便于对比不同IMS的分离性能,通常采用离子峰位置与其半高全宽的比值所定义的分辨能力作为替代[1]。DTIMS中,离子在均匀静电场的驱动下线性迁移,离子迁移时间td、离子迁移率倒数1/K及离子碰撞截面ΩD互成比例关系。选择任一尺度进行计算,均可得到一致的分辨能力和分辨率。对于具有非线性离子迁移机制的IMS,如TWIMS、TIMS等,由于1/K、ΩD与td不成线性关系,只有使用1/K或ΩD计算得到的分辨能力才能反映仪器的真实分离性能。目前,生物大分子构象分析领域普遍采用基于离子碰撞截面ΩD计算得到的分辨能力比对不同仪器的性能,示于式(2)[32]。
(2)
根据Shvartsburg等[29]定义,分辨能力超过80即为高分辨IMS,这是目前大多数商品化单机IMS所能达到的最高分辨能力。分辨能力超过200则为超高分辨IMS,可以使峰高相同、碰撞截面ΩD相差仅1%的2个谱峰实现基线分离[33]。目前,IMS技术中仅DTIMS、TWIMS、TIMS能够实现超高分辨。
本文将从超高分辨离子迁移谱的原理及技术,分辨能力的决定因素,优缺点,以及在生物分子构象分析中的应用及存在的问题和未来的发展趋势进行综述。
1 超高分辨离子迁移谱技术
1.1 迁移时间离子迁移谱(DTIMS)
DTIMS是最早出现的IMS技术。1930年,Tyndall与Powell[34]以及Bradbury与Nielsen[2]采用DTIMS研究气相离子迁移率K。1970年出现的世界首款军用化学战剂检测仪(CAM)[12]和离子迁移谱-质谱联用仪(IMS-MS)[3]也基于DTIMS。样品气体进入DTIMS后,形成的连续离子流会被周期开启的离子门斩切成离子团并注入长度L、电场E均匀的离子迁移区内,迁移率K不同的离子先后到达离子检测极,最终形成离子信号强度I对应离子迁移时间td的谱图,示于图1。其中,离子迁移率K可以通过测量离子迁移时间td直接得到,示于公式(3)。结合公式(1),可进一步获得离子碰撞截面ΩD,无需任何标定。目前,特殊设计的DTIMS可以将标准物离子迁移率K的测量精度控制在±0.1%以内,作为常规仪器中目标物离子迁移率K测量的参考标准[35]。
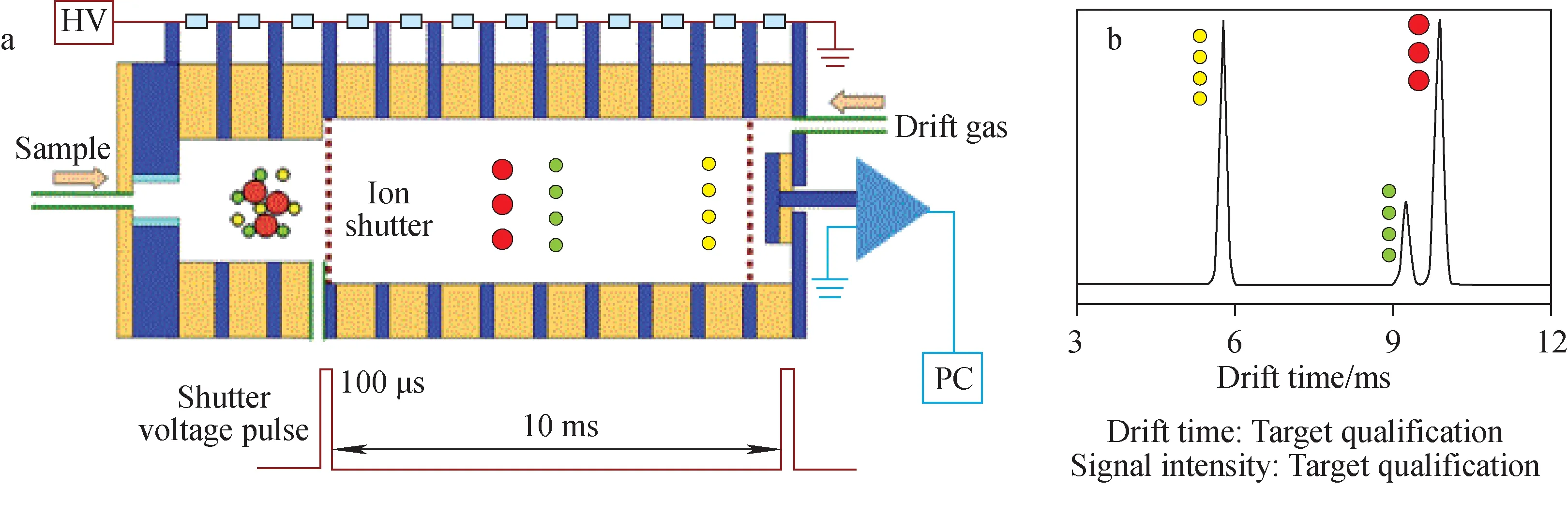
图1 迁移时间离子迁移谱结构(a)及离子分离原理图(b)Fig.1 Structure of drift time ion mobility spectrum (a) and the principle of ion separation (b)
(3)
DTIMS通过单次测量(约10 ms)可获得离子迁移率K全谱,并可通过多谱图平均提高信噪比进而降低检测限。但当DTIMS检测离子迁移率K差异较大的不同离子时,要求离子门具有极低的离子歧视效应,以消除离子迁移率K对离子团的初始时域宽度的影响[26,36]。
影响DTIMS分辨能力的因素有很多,如1) 离子团的扩散展宽;2) 离子团注入的初始时间宽度;3) 放大器响应畸变;4) 离子团之间的库仑斥力等。目前,已有较完善的理论模型对上述因素进行综合分析[37]。其中,普遍认为1)和2)是主要的影响因素,由离子迁移时间td、离子团注入的初始时间宽度tg、扩散系数D以及离子迁移速度vd,可以计算得到条件分辨率Rp,示于式(4)[38]。由于其他类型IMS仅考虑离子运动对分辨能力的影响,因此,其分辨能力的理论模型将仅考虑离子团扩散展宽的制约。
(4)
DTIMS既可以在大气压条件下工作(AP-DTIMS),也可以在低气压条件下工作(LP-DTIMS)。在大气压条件下,AP-DTIMS可与高效大气压化学电离源直接联用以获得高离子产率和灵敏度。另外,由于中性分子数密度N很高,即使施加较高的迁移区电压U,AP-DTIMS内约化迁移电场E/N仍很低(小于5 Td),并且远低于击穿极限。从式(4)可以看出,通过优化离子团注入效果可以提高分辨能力。Du等[39]通过建立BN型离子门的三区理论,示于图2,发现增加离子门的关门电压可以增大BNG后的电场强度,对注入离子团的空间压缩效应随之增强,即离子有效注入宽度t′g减小,对Cl-的分辨能力Rp由18增至33。在此基础上,Du等[40]通过在BN型离子门后放置1片金属栅网,使关门电压对门后电压的影响局限于BNG和该栅网之间,从而达到进一步增强BNG后电场的目的,使分辨能力进一步提高至60。但在BNG中,随着离子门的关门电压升高,离子团压缩效应增强,清空区也会增大,对离子迁移率的歧视效应增强。
Chen等[26]基于TP型离子门,通过提高2个栅网之间的电势差,提高了TPG的离子注入效率,从而在极窄的tg内保证极低的迁移率歧视效应,示于图3。在长9.65 cm的AP-DTIMS中通过提高关门电势差,将DMMP的分辨能力提升至100左右,并实现2个迁移率极相近(ΔK=0.04 cm2/(V·s))谱峰的基线分离。

注:a.BN型离子门的SIMION模型;b.BN门后的三区划分示意图;c.图a中x1=20 mm及x2=21 mm处的三区Ey特征线图2 BN型离子门周围的电场分布特征及离子门关门时的三区电场划分[39]Fig.2 Characteristics of electric field distribution around BN type ion gate and the three-zone division when ion gate was closed[39]

图3 通过增强TPG栅网之间电势差实现丙酮离子峰的分离[26]Fig.3 Separation of acetone ion peaks by enhancing potential difference between TPG grids[26]
在此基础上,Chen等[41]继续发展了一种离子双场时空压缩技术,通过在TPG后4 mm处放置1片金属栅网,改变离子门的控制模式,将注入的离子团进行连续两级压缩,从而将离子门性能提升至2倍左右,保证较高灵敏度的同时实现较宽迁移率范围(0.644~2.032 cm2/(V·s))内的高分辨,示于图4。
图4 双场压缩离子门模式下,各个时间段内离子的注入及压缩过程示意图[41]Fig.4 Elelctric potential in each state and the schemetic of the ion injection and compression process under dual-compression ion gate mode[41]
在保证离子门控注入和离子检测极具超快响应速度的前提下,可以通过提高迁移区电压U来优化AP-DTIMS的分辨能力。电压U与迁移区电场E、长度L直接相关,即U=EL,仅受电源实际输出能力的限制。Kirk等[33]通过在迁移区长15 cm的AP-DTIMS上施加25 kV高压,将单电荷小离子(如DMMP、苯、甲苯、丙酮)的分辨能力提升至250~260,是目前报道的DTIMS所能达到的最高分辨能力。另外,迁移区电压U较低时,增大离子价态z可以提高分辨能力。Clemmer等[42]通过在迁移区长63 cm的AP-DTIMS上施加10 kV高压,对单电荷C60团簇离子的分辨能力达到172,而负4价有机六聚体离子的分辨能力则达到240。
LP-DTIMS与质谱联用,可简化离子传输区域的匹配设计,还可以使用前置离子漏斗或离子阱进行离子预存储或选质。虽然二者联用有诸多优势,但仍无法获得类似AP-DTIMS的超高分辨能力,因为在几Torr气压范围内,中性分子数密度N极低,迁移区电压U不能无限制的提高。一方面,受到实验过程可容许E/N最大值的约束;另一方面,受到迁移区击穿极限E/N值的约束。此时,迁移区电压U可表示为迁移区最大场强Emax与迁移区长度L的乘积,即U=EmaxL。在不超过迁移区击穿极限的前提下提高E/N值,既可以获得更高的分辨能力,还可以对离子进行加热,进而研究离子迁移率K随E/N的变化规律。Kirk等[36]将LP-DTIMS与三态离子门控注入技术相结合,达到了消除迁移率歧视的效果,在长度30 cm、工作气压20 mbar的迁移区内施加120 Td的E/N,对水杨酸甲酯单价离子的分辨能力可达到140。基于此,Maria等[43]进一步研究了E/N在20~115 Td之间变化时,H3O+(H2O)n、NO+(H2O)n、O2+(H2O)n等离子迁移率K的变化规律。当实验测试要求较低的E/N值时,LP-DTIMS普遍采用长度1 m以上的迁移区来获得高分辨能力。Bowers等[44]发展了一种IMS-MS,采用迁移区长2 m、工作气压为15~20 mbar的LP-DTIMS,在5 kV迁移区电压下对血管紧张素Ⅱ正二价离子的分辨能力达到109。
目前,AP-DTIMS和LP-DTIMS均已经用于商品化IMS-MS中。例如,美国Agilent公司[45]推出的IM-QTOF 6560采用工作气压5 mbar、迁移区长80 cm的LP-DTIMS,其分辨能力仅80左右;瑞士Tofwerk公司[46]推出的IMS-TOF采用气压可调的AP-DTIMS,在离子复用模式下,可以实现约200的分辨能力;美国Excellims公司[47]推出基于AP-DTIMS的MA3100,其可与商品化超高分辨质谱仪(如LTQ-Orbitrap等)直接联用。
1.2 行波场离子迁移谱(TWIMS)
DTIMS的分离性能受到迁移区可施加最高电压U的限制,为了克服LP-DTIMS因这一限制无法获得超高分辨能力的问题,开始出现低气压条件下通过延长离子飞行距离来获得超高分辨的技术。
首先出现的是TWIMS,由Giles等[27]于2004年提出。美国Waters公司于2006年推出了首款商品化TWIMS仪器[27](Synapt HDMS Systems),后于2011年和2013年又推出了2款第二代TWIMS仪器[48](Synapt G2-S和Synapt G2-Si)。TWIMS迁移区由环状电极同轴叠合构成,在mbar气压范围内工作,示于图5。一方面,迁移区内相邻环状电极按照周期交替方式分别施加正、负射频电压[49],对迁移区内轴向迁移的离子进行径向约束聚焦;另一方面,迁移区内仅一小部分环状电极上施加沿迁移区轴向逐环向前推进的脉冲电压,于迁移区内形成幅度和波速可控的行波电场,驱动离子在行波电场中不断翻越波峰和波谷做冲浪运动。迁移率K越小,离子跟随行波电场前进的能力越差,迁移时间越长。由于离子在行波电场中做冲浪运动,离子迁移速度与迁移率K不再呈线性相关,并会受到行波电场类型及其电场强度分布特征的影响[50]。在理想的三角波行波电场中,离子迁移速度与迁移率K2相关;假定电场的波速正好能够驱动最大迁移率Kmax的离子随波迁移,离子迁移时间td,TW可由式(5)得到[50]。进一步由碰撞截面ΩD计算得到的分辨能力Rp,TW可表示为式(6)[50-51],其中e/16ln(2)kB是常量,可表示为C。

图5 行波场离子迁移谱中离子分离原理[27]Fig.5 Ion separation principle of traveling wave ion mobility spectrum[27]
(5)
(6)
在Emax、L等参数相同时,TWIMS分辨能力可以达到DTIMS的2倍。不同于DTIMS,TWIMS迁移区电压UTW与迁移区内行波场的波数m有关,即UTW=EmaxL/2m。当行波宽度固定时,UTW与迁移区长度L无关,因此,TWIMS可以在固定迁移区电压UTW下任意改变迁移区长度L,从而获得超高分辨能力。
对于第一代和第二代商品化TWIMS仪器,其分辨能力通常较低,约10[27]~40[48]。近几年,出现了2种通过延长离子飞行路径获得超高分辨TWIMS的技术。第1种技术采用循环路径的环形离子迁移谱(Cyclic-IMS),由Giles等[52]于2014年首次提出,Waters公司于2019年推出了基于该技术的最新款Select Series Cyclic IMS仪器[53]。Cyclic-IMS在mbar气压条件下工作,环形轨道上离子输入和输出共用1个端口。离子进入Cyclic-IMS后,可以在行波电场的驱动下进行多圈飞行,离子飞行路径正比于离子飞行圈数n,即nL,因此Cyclic-IMS的分辨能力随着n1/2增大而增大。根据Giles等[53]最近的报道,当离子飞行圈数达到100圈时,离子的飞行路径可达98 m,单电荷肽SDGRG和GRGDS的分辨能力可达750。射频聚焦电场可以使离子在较长的飞行距离下仍保持10%以上的离子传输效率。在Cyclic-IMS中,迁移率K分布较宽时,速度快的离子会与速度慢的离子谱图重叠。因此,通常缩减迁移率K的范围来保证离子具有相同的飞行圈数。
第2种技术被称为无损离子传输结构(SLIM),由Smith等[54]于2014年首次报道。最近,该技术已被MOBILion公司商品化。SLIM由蚀刻在PCB板上的平板条状电极构成,加工方便。SLIM在mbar气压下工作,通过射频聚焦电场的辅助,离子可以在SLIM中稳定存储数小时以上[55],并且能够在长达1 km的距离上进行近似无损传输[56]。SLIM内还可以设置离子操控结构,控制离子进行90°折弯传输[57]、同分离层内不同传输路径间切换[57]、不同分离层间切换[58]等,在有限的结构尺寸内构筑超长的离子飞行通道。例如,单分离层SLIM的离子飞行通道已从最初的44 cm[59]延长到13.5 m[56]。Smiths等[56]通过控制离子在SLIM中13.5 m长的分离通道内进行40圈往复飞行,将离子有效飞行距离延长至540 m,获得离子的分辨能力高达1 860,是已有报道的最高水平。但是,由于行波电场强度有限,离子飞行时间的延长会造成谱峰展宽、信噪比降低,因此,SLIM需通过基于SLIM的离子漏斗注入[60]、SLIM内离子捕集[61]、SLIM内离子团压缩[61]等技术增大离子数密度,提高信噪比。
1.3 阱离子迁移谱(TIMS)
TIMS是一种通过利用对向施加的高速气流和直流电场对离子的共同作用,从而增长离子有效迁移距离的IMS技术,由Fernandez-Lima等[62]于2011年首次报道。最近,Bruker公司基于该技术推出了商品化仪器timsTOF fleX。TIMS的离子分离区内通过施加流速为vg的高速气流推动离子向着接收极运动,同时施加强度随分离区轴线位置线性变化、方向与气流相反的位置电场阻止离子的迁移,示于图6[63-64]。离子进入TIMS后,当电场驱动离子的迁移速度与气流速度达到平衡时,即vg=KE,离子停止运动。如此,迁移率K不同的离子将会静止在TIMS分离区轴线的不同位置,被分离和富集。缓慢降低位置电场平台区的强度(洗脱电场)Ee,分离区中的离子将按照迁移率K由小到大的顺序依次洗脱,进入离子检测极或质谱中进行检测。类似TWIMS,TIMS也在mbar气压范围工作,需要使用射频电场对分离区内的离子进行径向约束。不同的是,TIMS采用四等分结构的电极环,施加射频后形成的离子聚焦电场为四极场。

图6 阱离子迁移谱的结构(a)及离子分离原理图(b)Fig.6 Structure of trap ion mobility spectrum (a) and the diagram of ion separation principle (b)
TIMS的分析时间tmeas,TIMS与洗脱电场的扫描速度β及离子迁移率K的分布范围有关,示于式(7)[51]。相对于DTIMS和TWIMS,TIMS可以省去从离子注入至迁移率K最大离子到达检测极的这段死时间。在实际分析中,tmeas,TIMS还与离子洗脱传输过程中的非线性项有关,导致离子迁移率K和碰撞截面ΩD无法直接计算求得[65]。因此,TIMS通常需要标定才能获得准确的迁移率K及碰撞截面ΩD数值[66]。
(7)
TIMS的分辨能力Rp,TIMS可表示为式(8),增大vg或者降低β均可以提高Rp,TIMS。Park等[64,67]使用β=3 356 V/(m·s)的扫描速度对泛素蛋白7价离子的分辨能力达到295,而使用β=2 691 V/(m·s)的扫描速度对m/z1 822单价离子的分辨能力仅为228。然而,增大vg的同时需要保证Emax条件下Kmin的离子仍能够被捕集于离子分离区内,即vg=KminEmax;降低β的同时会造成tmeas,TIMS的延长及谱峰的展宽,进而引起谱峰信噪比的降低。为解决这些问题,一方面,可以在TIMS离子分离区的前端设置前级离子漏斗对离子源中的离子进行预存储,以提高离子利用效率[68];另一方面,可以采用非线性洗脱电场扫描的方法,仅对感兴趣的迁移率K范围进行高分辨扫描,从而降低分析时间。这种非线性扫描方法有助于TIMS与速度较慢的质谱(如FT-ICR)进行配接[69]。
(8)
2 超高分辨离子迁移谱的应用
对天然大分子进行结构解析以了解其结构与功能的关系,一直是质谱学中备受关注的领域之一。由于IMS可以在毫秒内通过质量m、价态z以及碰撞截面ΩD等因素的差异实现离子分离,因此IMS与质谱联用(IM-MS)可以使离子在m/z分离的基础上,根据CCS的差异进行二维分离,可用于分析脂质[70]、聚糖[71-72]以及蛋白质[73-75]等生物大分子的结构,以及分离质谱无法分离的异构体[76-77],在临床医学、环境分析以及异构体分离等领域有广泛应用。随着目标样品复杂性的增加,分辨率较低(R约70)的IMS已经无法满足分离构象差异(ΔCCS%约0.5%)生物大分子的需求[32],而超高分辨IMS可以帮助解决这一难题。此外,通过超高分辨IMS有助于获取高精确度的CCS值,以建立CCS标准数据库。
由于DTIMS中的离子处于低场(E/N<2 Td)环境中,并在均匀恒定的静电场驱动下进行线性迁移,离子的CCS值可以直接通过式(1)与式(3)中的迁移时间td转换得出。理论上,通过精准测量实验参数并控制测量误差,DTIM-MS无需标准物的校准即可得到最精确的CCS值[78]。而当DTIM-MS进行在线分析时(例如与LC或GC联用),则需通过已知CCS值的目标离子进行标定。Stow等[79]通过对比3个使用Agilent IM-QTOF 6560仪器的实验室检测氮气气氛下120种离子DTCCSN2结果的重现性,其相对标准偏差(RSD)仅为0.29%。Tim等[80]依据EURACHEM通过蒙特卡洛模拟得到步进场和固定场2种CCS值测量方法得出的实验结果,相对理论值的偏差分别为2.7%~4.6%、4.7%~9.1%。DTIM-MS可用于多类生物大分子的结构分离及其CCS值的精准测定。May等[81]通过分辨率约为60的IM-Q-TOF MS测定出季铵盐、脂质、多肽以及聚糖等4类大分子的CCS值,从而对比不同类别分子之间的结构差异,具有较高的检测精度(±0.5%)。Groessl等[82]通过分辨率为250的Tofwerk IMS-TOF分离CCS值不同的脂质异构体,并且测量了130个大分子的K及CCS值,测量精确度约为1.3%,有助于建立相应的CCS标准数据库。但目前DTIMS-MS无法实现分离构象更接近的生物大分子。
随着TWIMS中超高分辨cIM以及SLIM等技术的发展,TWIM-MS已经可以实现约1 860的分辨能力[56],这使其在构象分离领域有巨大的应用潜力。David等[83]通过增大cIM-MS的飞行圈数(n=4),使分辨率达到约240,根据CCS以及m/z对红藻细胞壁中提取的天然低聚糖混合物进行分离并推断其具体结构,进一步增大飞行圈数(n=58),分辨率可达约920,能够分离开2个K极为接近(ΔCCS%约0.05%)的同分异构体。Hollerbach等[84]通过总长约为43 m的多级TW-SLIM-TOF MS,实现2个反向肽离子(ΔCCS%约1.5%)的分辨率由190增至560,更重要的是,这种仪器结构可以在较大的迁移率检测范围(3.0~1.2 cm2/(V·s))内仍具有超高分辨的能力。由于TWIMS中施加的射频行波电压使其电场非线性,因此无法通过td直接得出CCS,需通过已知CCS值的标准物对目标离子进行校准标定[85],即TWCCS=DTCCS*μ1/2/z。一些影响因素将导致TWIM-MS测定CCS值的精确度较低[82],如校定CCS时使用与目标物结构相差较大的标准物[86],不同平台间漂气温度的差异以及TW振幅可能引起的离子过热等。Hinnenkamp等[87]分别通过DTIM-MS及TWIM-MS对120种小分子离子的CCS值进行测定,发现二者检测部分化合物CCS值的结果相差6.2%。Li等[88]通过TW-SLIM-TOF MS检测不同类大分子的CCS值,相对DTIMS的检测结果偏差为1%~2%。
正如1.3节所述,TIMS可以通过增大vg或降低β来实现超高分辨能力[89],其分辨率可达到DTIMS分辨率的3~8倍,并且由于TIMS具有优异的占空比,更易与MS正交[68]。TIMS-MS主要用于研究蛋白质分子的结构表征,Park等[90]通过增加vg至100 m/s,减小洗脱电压的扫描速度δ至10 V/m,对多肽离子的分辨率可增至约250。为扩大TIMS检测的迁移率范围,Fouque等[91]通过对TIMS的电极环进行重新设计,以增强对较小迁移率离子(约0.524 cm2/(V·s))的捕集效果,通过增大vg以及降低β使分辨率达到约400,从而实现对几类蛋白质大分子(约924 kDa)异构体的分离分析,并建立了22种蛋白质CCS标准数据库,其CCS测量结果的重复性以及与DTIMS的检测结果相关性(R2=0.998 9)均较高。与TWIM-MS相似,TIMS-MS要获得准确的CCS值,同样需要标准物对目标离子进行校准标定,而标定的过程不可避免的会带来一定的误差。Naylor等[92]通过将双栅PCBIMS与TIMS-TOF耦合,从而可以在同一平台上直接测定离子精确的CCS值,避免了因标定过程引起的测量偏差,二者测定标准物K0的偏差约为1%,与文献中K0的偏差约为5%。
3 总结与展望
近年来,伴随着分辨能力超过200的超高分辨IMS技术的快速发展,出现了多种高性能商品化IMS-MS仪器,这为复杂样品(特别是生物样品)中分子构象的分析提供了重要的方法和工具。目前,不同的超高分辨IMS技术仍具有自身的局限性。例如,尽管TIMS和SLIM技术能够将IMS的分辨力提高到接近1 000的水平,但其面临着分析时间长、迁移率K窗口窄、需要特殊标定、离子热化效应影响测量精度等问题。在未来发展中,基于不同超高分辨IMS技术串联实现各自优缺点互补,将成为超高分辨IMS技术的一个重要发展方向。
猜你喜欢
数学物理学报(2022年5期)2022-10-09
数学杂志(2022年2期)2022-09-27
中学生数理化(高中版.高考理化)(2021年11期)2022-01-18
成都信息工程大学学报(2018年1期)2018-05-31
新高考·高一物理(2016年7期)2017-01-23
浙江大学学报(工学版)(2016年2期)2016-06-05
中学生数理化·高二版(2016年9期)2016-05-14
烟草科技(2015年8期)2015-12-20
郑州大学学报(医学版)(2015年1期)2015-02-27
广西科技大学学报(2015年4期)2015-02-27
