
基于图卷积神经网络的串联质谱从头测序
2021-09-18 06:22牟长宁王海鹏周丕宇侯鑫行
计算机应用 2021年9期
牟长宁,王海鹏,周丕宇,侯鑫行
(山东理工大学计算机科学与技术学院,山东淄博 255000)
(*通信作者电子邮箱hpwang@sdut.edu.cn)
0 引言
基于串联质谱的蛋白质组学是生命科学研究的重要组成部分,近年来在探索细胞机制、疾病进程以及基因型和表型之间关系等研究上取得了巨大进展[1]。基于串联质谱的蛋白质测序主流的方法是蛋白质数据库搜索,常用工具有Mascot[2]、Comet[3]、MaxQuant[4]、pFind[5-6]等。该方法需要参考已有数据库检索候选肽序列,因此在未知生物蛋白、单克隆抗体测序等研究上失去优势。另一种鉴定方法是从头测序,该方法能够直接从串联质谱中推断出氨基酸序列,无需数据库作为参考,在鉴定未知生物肽序列上具有不可替代的作用。过去20 年间从头测序方法进步显著,应用较为广泛的方案是基于图论的思想,将质谱转化为谱峰关系图(spectrum graph),谱峰作为谱图中的顶点,如果谱峰与谱峰之间的距离等于一个或者两个氨基酸残基的分子量,则两个峰之间用一条边相连;通过搜索图中起始点到结束点的最优路径得到产生这个质谱的候选肽序列。代表性工作包括:2003 年Ma 等[7]发表的PEAKS,通过预处理步骤(图谱噪声过滤和图谱峰聚合)创建谱图并用动态规划算法来生成候选肽序列;2005年Frank等[8]发表了针对碰撞诱导裂解(Collision-Induced Dissociation,CID)质谱的PepNovo 算法,提出了一种基于概率网络模型的候选肽序列评分方法;2010 年Chi 等[9]发表的pNovo,使用带剪枝的深度优先搜索有效提升了在高能碰撞裂解(Higher-energy Collision Dissociation,HCD)质谱数据上的从头测序性能;随后同一团队,在pNovo 基础上开发了同时使用HCD 和电子转运裂解(Electron Transfer Dissociation,ETD)数据的从头测序方法pNovo+[10],以及针对翻译后修饰肽鉴定的OpenpNovo[11],并在2019年发表了pNovo3[12],将理论质谱预测用于候选肽重排序。另一类从头测序方法则是基于机器学习和深度学习技术。2005年,NovoHMM算法[13]提出使用隐马尔可夫模型解决从头测序问题;2015 年,Novor[14]使用决策树模型分别为碎片离子和氨基酸残基进行打分,结合动态规划推导肽序列;基于深度学习的DeepNovo[15],通过基于卷积神经网络(Convolutional Neural Network,CNN)的Ion-CNN 和Spectrum-CNN,以及长短期记忆(Long Short-Term Memory,LSTM)网络模型融合的方式对肽序列进行预测。随着从头测序方法的改进,测序精度不断得到提升,然而由于质谱仪中肽不完全碎裂等因素,导致质谱中碎片离子的覆盖率较低,重要b离子或y离子峰丢失,大量噪声干扰峰难以通过约束条件彻底清除,诸多因素致使从头测序的精度仍然较低,严重制约了从头测序在蛋白质组数据分析中的应用。因此提升肽段从头测序准确性,对蛋白质组学研究具有重要意义。
在蛋白质组学中,深度学习方法已经应用到了预测肽段保留时间、理论质谱预测、翻译后修饰、从头测序、蛋白质结构预测等多个任务中[16-17]。深度学习的蓬勃发展,为质谱数据分析不断提供新的方案启示。本文在经典的谱峰关系图方法基础上,提出了一种基于图卷积神经网络(Graph Convolutional neural Network,GCN)的从头测序方法denovo-GCN。该方法直接使用质谱数据作为输入,简化中间数据约束处理过程,在谱峰关系图结构上按照碎裂位点为每个谱峰构造特征表示。通过在大规模数据上的训练优化,能够有效提升从头测序的准确性。
1 denovo-GCN模型
1.1 图卷积神经网络与从头测序
图卷积神经网络以其在图数据上的强大建模能力,在知识图谱、社交网络等众多领域得到了应用[18]。Kipf 等[19]对ChebNet[20]进行了简化,提出了一种更加简单的模型GCN,它相当于对一阶切比雪夫卷积的再近似,降低了计算复杂度,并且可以通过堆叠多个GCN 扩大图卷积神经网络的感受野,实用性大大增强。GCN模型结构表述为式(1):
其中:=A+I,A是图的邻接矩阵,包含了节点之间的连接信息;I是单位矩阵,加上I后得到的包含了自身节点和邻接点的信息是顶点的度矩阵是激活函数;H(l) ∈Rn×m是第l层的激活矩阵;H(0)=X,X是由各节点特征向量xi组成的特征矩阵。随后注意机制、序列模型等也用于图中节点权重的计算,图卷积神经网络呈现出多样化的发展。
从头测序过程可以类比为语言翻译或者图像描述,最终目的是得到一个映射原始数据的序列表示。不同之处在于后者的原始数据是规则欧氏空间数据,而质谱数据是一组谱峰质荷比及其强度的数据对组成的集合。在基于图论的从头测序中,谱峰关系是由谱峰之间的距离来计算,形成谱峰连接图。这种质谱数据图结构化的表示方法与针对图结构数据的图卷积神经网络十分契合。谱峰节点的特征则可以通过枚举碎裂位点产生的离子与各谱峰的距离关系表示,借助图卷积神经网络的训练学习能力将符合条件的谱峰点与干扰峰进行区分,预测当前位置的氨基酸身份,逐步实现氨基酸序列的推理。
1.2 图的构建
在质谱数据上,使用图卷积神经网络的首要任务是构建谱峰连接图。质谱数据中的关键信息包括母离子的质荷比、肽所带电荷、谱峰。谱峰是碎片离子质荷比及其强度组成的数据对,将谱峰强度值按照同一质谱中最大强度值归一化得到相对强度,相对强度最大值为1。单个质谱可以直观表示为质荷比和相对强度的柱状图,x轴代表质荷比,y轴代表强度。若谱峰与谱峰之间的距离与一个氨基酸残基的分子量的差值在设定误差范围内,则两个谱峰之间建立一条边。在构建谱图前,需向原始谱图添加序列端点的谱峰,分别为一个电荷(M(proton))、一个水分子量(M(H2O))、1 电荷肽的分子量(M(peptide))、肽失去一个水的分子量(M(peptide)-M(H2O))四个谱峰点,相对强度皆设置为1。设S=为谱(npeaks为谱峰数量),SA为峰与峰之间的差值矩阵,MASS_AA=(n=23,代表20 氨基酸残基和3 种修饰后的氨基酸残基)为氨基酸残基质量集合,计算邻接矩阵的过程用式(2)~(5)表示:

由式(2)计算谱峰差值矩阵绝对值与每个氨基酸残基的误差矩阵,如果误差在给定ε内则将相应元素标记为1,若超出范围则标记为0,然后将所有矩阵相加得到当前谱的邻接矩阵;加入相同维度的单位矩阵作为节点自身的信息,避免构图时谱峰为孤立峰,即谱中没有相邻位点产生的同类型离子谱峰,导致不存在边与之相连造成信息丢失;再计算度矩阵并对邻接矩阵进行归一化。
将质谱数据处理成图结构化数据是denovo-GCN 与DeepNovo处理质谱数据的不同之处,在DeepNovo中将串联质谱数据对应成规则的欧氏数据,质荷比维度的数据按照质量精度0.01 Da(Dalton)进行扩展:假设谱中的最大质荷比为1 500.00 Da,整个谱离散化为150 000 个刻度,再将每个谱峰相对强度填入离散化后的刻度位置,卷积提取特征。而在denovo-GCN 中,谱峰之间的关系直接计算确定,不需要通过深度学习模型来学习这种关键信息。
1.3 谱峰特征构建
denovo-GCN 的另一个关键在于为质谱中的每一个谱峰建立特征。由于串联质谱数据的特殊性,很难在只使用一组离子质荷比和谱峰强度数据条件下推断出序列信息,因此必须利用肽碎裂产生的离子类型设计特征。肽段在HCD 模式下常见的碎片离子类型有b、y、b2+、y2+、b-H2O、y-H2O、b-NH3、y-NH3、a、a2+、a-H2O、a-NH3等[21],在计算得到b离子或者y离子质荷比后便可根据母离子质荷比计算同一断裂位点的其他离子质荷比。在模型中,设定了26种符号标记分别代表20种氨基酸残基、3 种修饰后的氨基酸残基、3 种特殊的标记(start、end、pad)。特征可以看作是当前碎裂位点产生的离子与谱峰的距离差值,构建过程如式(6)~(8):


设ntoken为设定标记的个数,nions为使用的离子类型的种类,计算得到的理论质荷比矩阵为Mt大小为(1,ntoken×nions),将其按第一维度复制得到Mt'(npeaks,ntoken×nions()npeaks为谱峰数量);将当前谱峰矩阵Mo(npeaks,1),按第二维度复制得到Mo'大小同样为(npeaks,ntoken×nions),由式(6)计算谱峰与理论离子的误差矩阵E,然后通过指数运算将误差值缩放到区间(0,1)内,⊕代表将谱峰的相对强度Intensity(npeaks,1)拼接到E,形成了最终的特征矩阵F。
1.4 denovo-GCN的模型构建
denovo-GCN 的模型如图1 所示:由质谱数据分别计算谱图邻接矩阵和初始特征矩阵。使用GCN 对质谱数据进行特征提取,按照谱峰的维度加和并使用Leaky ReLU 激活函数进行激活,再使用全连接层输出,得到氨基酸类型的概率,输出当前条件下的氨基酸身份。
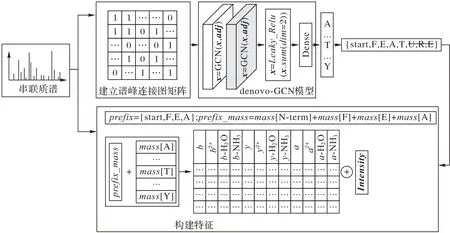
图1 denovo-GCN模型Fig.1 denovo-GCN model
新预测的氨基酸加入到序列后,计算下一个位点的特征矩阵,直至出现结束标记或者达到设定的序列最大长度。模型各层的参数大小设置如表1 所示,其中ntoken为设定标记的数目,nions为使用的离子类型的数目。训练时标注肽序列中的每一个氨基酸作为标记,依次进行批训练,初始学习率为0.001,根据模型训练评价自适应调整学习率,最低学习率设置为10-5。由于肽序列中氨基酸出现的频率差别很大,特别是带有修饰的氨基酸残基占比更少,因此在训练时使用了Focal Loss 函数计算损失,该函数最初用于解决目标检测中类别不平衡问题[22]。

表1 denovo-GCN模型中各层的参数Tab.1 Parameters of each layer in denovo-GCN
1.5 评价指标
通常从肽水平和氨基酸水平上评价从头测序结果[12-15]。肽水平召回率和精确率分别为完全预测正确的肽序列占测试数据中所有肽序列的比例和接受的测序结果中肽序列总数的比例,氨基酸水平召回率和精确率分别为预测正确的氨基酸总数分别占测试数据中氨基酸总数的比例和接受的测序结果中氨基酸总数的比例。在氨基酸水平上,从N 端或C 端开始对应位置预测的氨基酸与标注一致则为正确,对于分子量相同的亮氨酸(Leucine,L)和异亮氨酸(Isoleucine,I),在同一位置时认为预测正确。
2 实验与结果分析
2.1 数据集和模型结构优化
本文在ProteomeTools1(ID:PXD004732)数据集[23]上进行了模型的训练和测试,确定了模型的结构、离子类型组合和采用的谱峰数量。该数据集来自人工合成蛋白质数据集,从proteomeXchange 蛋白质数据库中获得,根据MaxQuant搜索结果以得分score≥100、PIF≥0.7(Precursor Intensity Fraction)过滤选取高质量的肽谱匹配数据,最终得到204 996 条标注数据,并在实验时以8∶1∶1 的比例随机划分训练集、测试集、验证集,集合划分时相互不存在交集。实验中构建谱峰关系图时使用的质量误差ε为0.02 Da。
不同层数的GCN 模型效果根据具体应用会有所差异。本节实验设置最大谱峰数量为500,离子类型为12种,GCN的hidden size为256,实验结果如表2 所示:实验中采用2 层GCN的模型比使用1 层和3 层的模型在肽水平的召回率分别高出2.5个百分点和1.2个百分点,比直接使用全连接网络高出了2.9个百分点,4层的模型与3层的模型效果基本一致;各组模型氨基酸的召回率在91.19%至92.19%。在氨基酸水平召回率相近的条件下,GCN模型明显提高了肽水平的召回率,并在使用2 层GCN 结构时获得最高召回率。因此,后续实验皆采用2层的GCN结构。

表2 不同GCN层数模型的召回率对比 单位:%Tab.2 Comparison of recall by different GCN layers’models unit:%
2.2 碎片离子类型的选择
肽段在高能碰撞裂解(HCD)碎裂模式下,主要产生b/y离子及带二电荷的常规离子,也会产生常规离子失去水分子和失去氨分子的中性丢失离子,以及a型离子。为了测试不同离子类型组合对模型的影响,以b/y离子组合为基础,测试了加入不同离子类型后的表现,该部分实验谱峰数量设置为500,实验结果如表3所示。在加入2电荷的b/y离子后肽召回率比只使用1电荷b/y离子时提升了16.0个百分点,氨基酸水平提升了7.3 个百分点。b、y、b2+、y2+在测序中起着关键作用,这与HCD 谱中关键离子为b/y离子的特性是一致的。当模型中继续加入b/y离子的中性丢失离子(b-H2O、y-H2O、b-NH3、y-NH3)时,肽的召回率比使用4种常规离子增加了3.7个百分点,氨基酸水平增加了1.3 个百分点;在加入a型离子及其中性丢失离子(a、a2+、a-H2O、a-NH3)后模型肽水平召回率再次提升了2.1 个百分点。当离子从4 种增加到12 种时,氨基酸水平的召回率只提升了1.9个百分点,但肽的召回率提升了5.7个百分点。这说明,额外增加的8 种离子提供了更多测序信息。当谱中没有出现某一碎裂位点的常规离子,但存在对应中性丢失的离子峰时,同样可以为该处氨基酸身份的鉴定提供依据。因此丰富的离子类型组合可以提升测序的准确度。

表3 不同离子类型组合的召回率对比 单位:%Tab.3 Comparison of recall by different combinations of ion types unit:%
2.3 谱峰数量的影响
除离子类型组合会影响模型,每个谱采用的谱峰数量也会对模型产生影响。质谱中存在大量低丰度的离子峰和噪声峰,基于图论等其他从头测序方法中会先对实验谱消除一部分同位素峰和相对强度过低的峰。在denovo-GCN 中采用简便的方式,保留相对强度在给定排名内的谱峰。为了验证谱峰数量的影响,实验以每个谱选取64 个峰为起始,每次实验递增64 个峰,最大峰数为640,实验结果如图2 所示。首先统计测序时使用的谱峰数量(used peaks)占全部数据的谱峰数量(total peaks)的变化曲线。当选取256 个谱峰进行实验时,实验中用到的谱峰数量占总数据的70.62%,此时谱中的关键峰基本纳入到了考虑范围内;选取谱峰数量为384 时占比达到89.63%;选取谱峰数量为512 时占比达到97.39%,接近全部数据。在谱峰数超过256 个时,肽召回率均值为77.84%,模型的准确率趋于稳定。当使用384 个谱峰时,基本将大部分谱峰纳入到测序中,且使用384 个谱峰时训练时间比使用512 个谱峰时减少了1/3,若考虑使用全部谱峰时可选择512个谱峰。

图2 肽水平的召回率随谱峰数量的变化曲线Fig.2 Curve of peptide-level recall varying with number of spectral peaks
2.4 不同测序方法在ProteomeTools1数据上的对比
在确定了模型结构、离子类型组合、谱峰数量后在ProteomeTools1 数据集上对denovo-GCN(12 种离子类型,384个谱峰)、DeepNovo(version 0.0.1)、pNovo(version 3.1.3)、Novor(DeNovoGUI version 1.9.6)进行了测试。上述工具给出了预测肽序列的得分,将最终结果按照得分从小到大排序,给定分数t,计算肽水平的精确率(得分至少为t的实际正确肽数量/得分至少为t的肽数量)和召回率(得分至少为t的实际正确肽数量/数据中总的肽数量),画出肽水平上的精确率-召回率(Precision-Recall,PR)曲线如图3所示。
从图3 可看出,denovo-GCN 的曲线明显高于DeepNovo、Novor 的曲线,召回率在区间[0,0.5]内与pNovo 的曲线有重合的部分,召回率超过0.5时明显高于pNovo。再分别计算各PR曲线下的面积,denovo-GCN 为0.731 8,DeepNovo 为0.613 8,pNovo为0.619 2,Novor为0.518 1。denovo-GCN 在同一数据上的测序性能要优于其他三种工具。
2.5 不同物种数据的交叉对比
在实际应用中,从头测序更多的是解决未知物种蛋白的测序。因此,为了进一步验证denovo-GCN 的测序表现,本节采用了DeepNovo 中的9 个HCD 数据集,进行物种间的交叉对比实验,数据信息如表4所示。

表4 9个HCD数据集信息Tab.4 Information of 9 HCD datasets
每次使用其中的8 个数据集混合划分训练集、验证集进行模型训练,集合之间不存在肽序列交集,未参与模型训练的1 个物种数据作为测试集。用相同的数据分别训练DeepNovo和denovo-GCN(12 种离子类型,384 个谱峰),Novor 和pNovo直接使用其提供的软件进行测序,测试结果如图4所示。

图4 denovo-GCN、Novor、pNovo、DeepNovo在9个HCD数据集上的实验结果对比Fig.4 Experimental result comparison of denovo-GCN,Novor,pNovo,DeepNovo on 9 HCD datasets
图4(a)是不同工具间氨基酸水平的召回率对比,denovo-GCN 比Novor 高出6.2~32.7 个百分点,比pNovo 高出7.6~14.9 个百分点,比DeepNovo 高出4.3~9.9 个百分点。图4(b)在不同工具上氨基酸水平的精确率对比,denovo-GCN 比Novor 高出3.8~31.1 个百分点,比DeepNovo 高出4.1~10.0 个百 分 点,而pNovo 在H.sapiens 数 据、M.musculus 数 据、Candidatus 数据上比denovo-GCN 的精确率高出6.1 个百分点、3.7 个百分点、2.4 个百分点,其余数据上denovo-GCN 比pNovo 高出2.2~4.9 个百分点。图4(c)在肽水平上不同工具的召回率对比,denovo-GCN 的肽的召回率比Novor 的高出9.8~21.1 个百分点,比pNovo 高出4.0~13.0 个百分点,比DeepNovo 高出2.1~10.7 个百分点。综上实验结果denovo-GCN相较于Novor、pNovo、DeepNovo,能够测得更多的氨基酸,并且能够转化成更多正确的肽序列,测序能力超过了其他三种工具。相较于DeepNovo的模型结构,denovo-GCN模型更为精简,使用图来表达谱峰之间关系并结合图卷积神经网络的方式比CNN和LSTM模型在串联质谱测序上更具优势。
对于表4的9个物种的测试数据在以ProteomeTools1数据训练的denovo-GCN、DeepNovo 模型上分别进行测试,并使用ProteomeToolsV2(ID:PXD010595)[24]人工合成肽的数据作为相似物种进行对比参照,得到的结果如表5 所示。在相似物种上两个模型表现都要好于非同类物种的表现,而非同类物种上由于蛋白质差异,测序效果存在一定差距。这两部分实验结果表明denovo-GCN 的测序能力优于DeepNovo、pNovo、Novor。
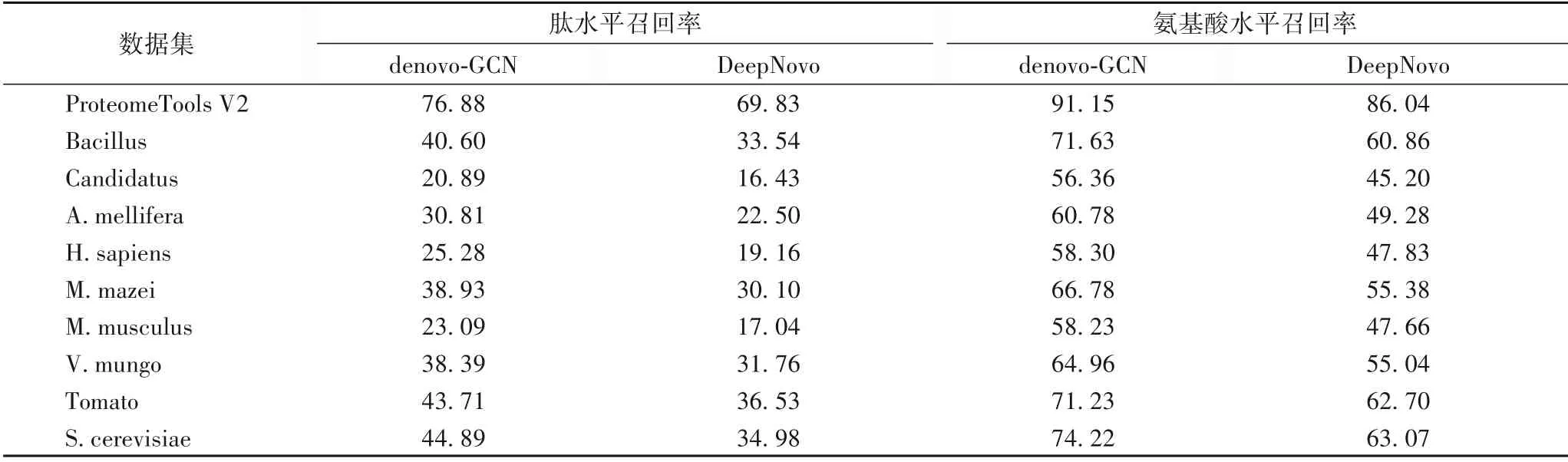
表5 ProteomeTools1数据集训练的模型在9个HCD数据集上的实验结果 单位:%Tab.5 Experimental result on 9 HCD datasets by the models trained on ProteomeTools1 dataset unit:%
2.6 denovo-GCN与pNovo预测结果及序列分析
pNovo 是基于图论的从头测序的代表,在几个测试数据上氨基酸召回率虽然低于DeepNovo,但肽的召回率却与之接近。为了查看预测序列中出现的错误肽序列,在ProteomeTools1 测试数据上pNovo、denovo-GCN 测序结果和数据库搜索结果之间的关系如图5 所示:两者有12 661 条数据测序结果相同,同时互有无法给出对方正确测序结果的数据,但denovo-GCN较pNovo多鉴定出了1 451条。
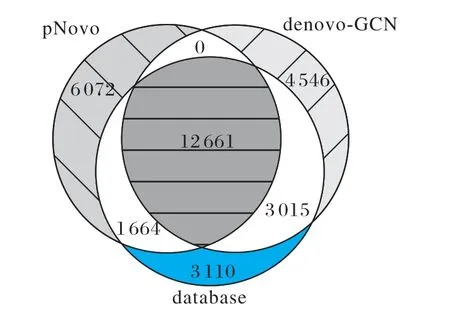
图5 pNovo、denovo-GCN、数据库搜索结果的文氏图Fig.5 Venn diagram of pNovo,denovo-GCN,database search results
对两者测序均为错误的结果进行分析,总结了测序时出现频率较高的3 种错误类型,示例如表6 所示:1)当串联质谱中的低质量区域,存在较多的亚胺离子和内部离子,而关键性的低质量常规离子峰与之不易区分甚至缺失,在构图时会出现多条互相连接的边,氨基酸位置难以确定;2)氨基酸残基存在单个氨基酸分子量等于两个小质量氨基酸之和或者两种不同氨基酸分子量之和两两相等的情况,谱中两端缺失了关键的b/y离子;3)在长序列谱或低质量谱中,离子峰更为复杂,谱峰可以对应多种氨基酸序列的组合,在测序时较难得出正确氨基酸组合。这也能够解释denovo-GCN 在不同物种数据实验中氨基酸的召回率能够达到60%以上,而肽序列的正确率却在25%~48%。解决上述问题最直接的方法是提升质谱仪输出数据的质量,而当前质谱数据条件下,解决上述问题的思路主要有两个:1)算法模型输出多个候选肽序列并进行再次打分,找出更优的序列;2)不断探索创新测序算法,从而提高肽序列的正确率。
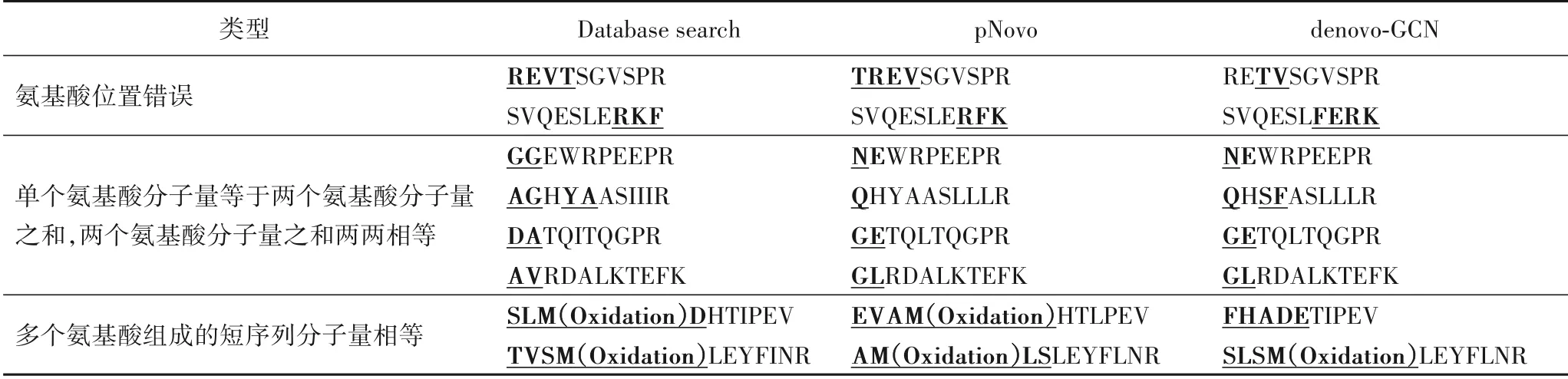
表6 pNovo与denovo-GCN结果中典型的序列错误示例Tab.6 Examples of typical sequence errors in pNovo and denovo-GCN results
3 结语
denovo-GCN 将质谱数据转化为图结构数据,根据肽碎裂产生的离子类型对每个谱峰点进行特征设计,将图卷积神经网络引入到从头测序任务中,提升了串联质谱测序的准确率,超过了基于图论的从头测序方法Novor、pNovo,以及基于CNN和LSTM 模型的DeepNovo。实验结果表明充分利用肽碎片离子类型,选择适当谱峰数量作为参数可以取得较为理想的效果。虽然denovo-GCN 实验中同数据集上可以达到数据库搜索结果70%的肽召回率,并且在不同物种测序上也好于其他工具,但不同物种数据的测试结果并未超过数据库结果的50%。denovo-GCN 的测序效果会受到训练数据的影响,可以通过扩大训练数据种类来消除部分影响。提升从头测序的准确性,仍是一项值得持续研究的课题,而另一方面,如何测定序列中修饰后的氨基酸类型也需要进一步研究。
猜你喜欢
中国人兽共患病学报(2022年9期)2022-10-19
信阳农林学院学报(2022年2期)2022-08-06
中国畜牧杂志(2022年6期)2022-06-13
现代仪器与医疗(2022年1期)2022-04-19
食品安全导刊(2021年21期)2021-08-30
湖南饲料(2021年3期)2021-07-28
现代仪器与医疗(2021年2期)2021-07-21
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
分析化学(2019年3期)2019-03-30
