
黑骨藤LC-MS定性分析及4种绿原酸一测多评分析方法
2021-09-13 14:16李晨阳赵喜玲
四川师范大学学报(自然科学版) 2021年5期
李 琪, 王 博, 李晨阳, 赵喜玲, 陈 坤, 张 宏*
(1.四川师范大学 生命科学学院,四川 成都610101; 2.四川师范大学 植物资源应用与开发研究所,四川 成都610101)
黑骨藤(Periploca forrestiiSchltr.)为萝藦科(Asclepiadaceae)杠柳属(Periploca Linn)植物,又名黑龙骨(HLG),曾用于治疗类风湿性关节炎和一些创伤[1].现代药理实验证明,黑骨藤具有抗氧化[2]、抗炎[3]、抗癌[4]、神经保护[5]等作用.然而,黑骨藤的化学成分复杂,各种化学成分的极性差异很大,这使得很难对其进行定性研究.目前,黑骨藤化学成分相关研究报道较多,成分包含挥发油、皂苷、杠柳苷等多种类型化合物,并已建立了HPLCQ-TOF-MS对其进行定量测定[6-7].近年来,一种新的、简单快速的技术——UPLC-Q-TOF-MS得到了广泛应用,它可提供关于特征分子离子和碎片离子的准确的质量信息,在代谢物的鉴定和结构解析中起着重要的作用[8-9].因此,本研究采用UPLC-QTOF-MS技术.
外标法是最常用的评价方法之一,由于一些对照品具有高成本、化学多样性和检测的不稳定性等因素,使其具有局限性[10-11].为了解决这一问题,王智民等[12]首次系统地提出了一测多评法(QAMS).中国药典(2015版)已报道此法适用于中药材的含量测定[13],其优点是每种分析物的含量可以独立测定,并且可以减少实验成本和检测时间[14-15].基于目前关于黑骨藤QAMS法的缺乏,本文采用了QAMS法,以隐绿原酸为内标物,分别计算其与绿原酸、异绿原酸C、新绿原酸的相对校正因子(RCF)而得出其含量,从而建立黑骨藤中4种绿原酸的一测多评分析方法,为黑骨藤的质量评价提供了一种快速、实用、有效的方法.
1 材料、试剂与方法
甲醇、乙醇(分析纯,成都市科龙化工试剂厂);乙腈(色谱纯,美国天地试剂公司);磷酸、甲酸、醋酸(色谱纯,成都市科龙化工试剂厂);硅胶(200~300目,青岛海浪硅胶干燥剂厂).绿原酸、新绿原酸(成都瑞恩思生物科技有限公司),隐绿原酸、杠柳毒苷、原花青素A1、原花青素A2、异绿原酸C(深圳市斯坦德合成有限公司).所有对照品经UPLC峰面积归一化法检测质量分数均大于98%.
1.3 色谱条件
1.3.1 UPLC-Q-TOF-MS色谱条件 色谱柱:Eclipse Plus-C18(100 mm×4.6 mm,1.8μm).流速:0.7 mL/min.柱温:30℃.进样量:5μL.检测波长:221 nm.流动相:流动相A(体积分数为0.1%的甲酸溶液);流动相B(体积分数为0.1%的甲酸乙腈).梯度洗脱程序:0~8.0 min,95%~85%A;8.0~12.0 min,85%~82%A;12.0~20.0 min,82%A;20.0~25.0 min,82%~80%A;25.0~40.0 min,80%~70%A;40.0~60.0 min,70%~5%A.质谱条件:正负离子扫描模式.扫描范围(m/z):100~1 500.雾化气(GS1):50 psi.雾化气(GS2):50 psi.气帘气(CUR):30 psi.离子源温度(TEM):-550℃,600℃.离子源电压(IS):-4 500 V、5 500 V.一级扫描:去簇电压(100 V);聚焦电压(10 V).二级扫描:采用IDA模式,CID能量为20、40和60 V.
1.3.2 QAMS法色谱条件 色谱柱:ZORAX SB C18(2.1 mm×100 mm,1.8μm).流速:0.3 mL/min.柱温:40℃.检测波长:325 nm.进样量:1μL.流动相A:甲醇;流动相B:体积分数为0.1%的磷酸水溶液(pH=3.0).梯度洗脱程序:0~10.0 min,5%~25%A;10.0~25.0 min,25%~40%A;25.0~30.0 min,40%~60%A.
1.4 混合对照品溶液的制备 称取新绿原酸(6.70 mg)、绿原酸(7.54 mg)、隐绿原酸(4.71 mg)和异绿原酸C(4.84 mg)、原花色素A1(4.25 mg)、原花色素A2(4.39 mg)和杠柳毒苷(4.48 mg)于10 mL容量瓶中,色谱甲醇溶解定容到刻度以制备混合标准溶液,再经0.22μm滤膜过滤后待用.
1.5 供试品溶液制备 称取已活化的硅胶200 g于1 L烧杯中,采用湿法装柱,加入600 mL石油醚搅拌均匀装柱.粉碎黑骨藤样品,过3号药典筛.称取50 g干粉于1 L锥形瓶中,按料液比1∶20,加入体积分数为50%的乙醇1 L,超声(功率200 W,频率40 kHz)提取30 min,提取温度50℃,提取2次.将2次提取液合并减压浓缩旋干至干粉.取5 g干粉于50 mL烧杯中,添加10 g硅胶,加入40 mL甲醇,于通风橱中待自然风干成细粉末后上样.依次用石油醚、V(石油醚)∶V(乙酸乙酯)=1∶1、乙酸乙酯、V(乙酸乙酯)∶V(甲醇)=1∶1、甲醇洗脱,最终收集乙酸乙酯片段和V(乙酸乙酯)∶V(甲醇)=1∶1片段.称取上述2个片段各10 mg加入5 mL甲醇于离心管中,超声处理(功率200 W,频率40 kHz)30 min,离心10 min(12 000 RPM,4℃).取上层清液转移到1.5 mL离心管中,200μL体积分数为50%的甲醇溶液溶解,经0.22μm微孔膜过滤后,供UPLC-Q-TOF-MS分析.
在QAMS方法建立中,将S1样品收集粉碎,过3号药典筛.称取1 g左右的干粉于50 mL锥形瓶中,按料液比1∶20,加入体积分数为50%的乙醇20 mL,超声提取30 min(功率200 W,频率40 kHz),提取温度50℃,提取1次,抽滤、减压浓缩旋至近干,色谱甲醇洗3次转移到5 mL容量瓶中,定容得黑骨藤供试品溶液.
从图2可看出,谷索的就位对吊索索力影响很小;脊索就位对不同索的索力影响不同;膜的安装将提高吊索的索力。
1.6 QAMS方法学考察
1.6.1 标准曲线及检测限、定量限 精密移取混合对照品溶液各0.3、0.5、0.8、1.0、2.0、4.0 mL,分别用色谱甲醇定容于10 mL容量瓶中.每个浓度平行测定3次,记录混合对照品的峰面积,以峰面积为纵坐标,溶液质量浓度为横坐标,建立隐绿原酸、异绿原酸C、新绿原酸、绿原酸的标准曲线,得线性方程、线性范围、相关系数.选取最低浓度对照品,采用逐级稀释方法,每次稀释2倍,按照色谱条件进行检测.借助信噪比,以S/N=3为检测限,以S/N=10为定量限.
1.6.2 精密度、重复性、稳定性和加标回收率 平行测定混合标准溶液和S1供试品溶液6次,以考察仪器精密度和样品精密度.选取6份1.000 g的S1号黑骨藤样品考察重复性,并在0、2、4、6、8、10、12、24和48 h对供试品溶液进行测定,以获得其稳定性.选取3份5.000 g的S1号黑骨藤样品,每份按药材与对照品质量比约1∶1的量加入对照品,根据(1)式[16]计算每种分析物的加标回收率.精密度、重复性、稳定性和加标回收率的相对标准偏差均用RSD表示.

1.7 RCF的测定及评价精密移取混合对照品溶液各0.3、0.5、0.8、1.0、2.0、4.0 mL,色谱甲醇定容于5 mL容量瓶,色谱甲醇定容至刻度,摇匀,过0.22 μm微孔滤膜.由于在一定的线性范围内,组分的浓度与检测器的响应成正比[10],本次实验以隐绿原酸为内参物,对每个浓度平行测定3次,分别运用(2)式[11]计算隐绿原酸相对于其他组分的相对校正因子(RCF),用f表示,并计算其相对标准偏差RSD.As为内参物峰面积,Ws为内参物质量,Ai为其他组分的峰面积,Wi为其他组分的质量.

分析物的浓度是影响RCF的主要参数之一,但RCF还受其他实验条件的影响[11].本次实验考察了不同色谱柱及其温度和不同进样量对RCF的影响以验证其稳定性.
1.8 待测成分色谱峰的定位待测成分色谱峰的准确定位是建立QAMS的前提和关键,主要有2种主要的峰定位方法,保留时间RT(retention time)和待测成分与内标物之间的相对保留时间RRT(relative retention time)[15].本实验以隐绿原酸与其他3种组分的相对保留时间值对黑骨藤中待测成分色谱峰进行定位.
1.9 外标法与一测多评法的比较分别用外标法和一测多评法计算S1~S8不同产地黑骨藤中隐绿原酸、异绿原酸C、新绿原酸、绿原酸的含量,基于t检验、相关系数R[17]、相对标准偏差RSD[18]和相对误差δ[19],验证一测多评法与外标法是否存在差异性.
2 结果
2.1 UPLC-Q-TOF-MS结果各洗脱片段UPLC分析结果如图1所示.

图1 硅胶柱层析洗脱色谱图Fig.1 Chromatogram of silica gel column chromatography elution
分析可知:石油醚片段和甲醇片段含有少量的峰,而在其他2个片段中含有大量的峰,且其中有的峰与石油醚片段和甲醇片段中的有所重复,表明大多数组分存在于乙酸乙酯片段和V(乙酸乙酯)∶V(甲醇)=1∶1片段中,故最终选择上述2个片段用于定性分析,它们的总离子流色谱图如图2所示,具体参数见表2.通过比较分子式、保留时间tR、准分子离子和产物离子,质量误差均在8.6×10-6以内,共分离鉴定出10种化合物.

图2 黑骨藤提取物总离子流色谱图Fig.2 Total ion chromatograms of the extracts from HLG

表2 黑骨藤提取物中被鉴定出的10种成分Tab.2 Identification of 10 components in HLG extracts
2.2 方法学考察结果回归分析的结果见表3,相关系数R≥0.999 5,表明所有组分均表现出良好的线性,回归方程可用于给定浓度范围内的定量分析.由表4可得RSD值均小于2.0%,表明本方法具有良好的准确性和重现性,稳定可行.
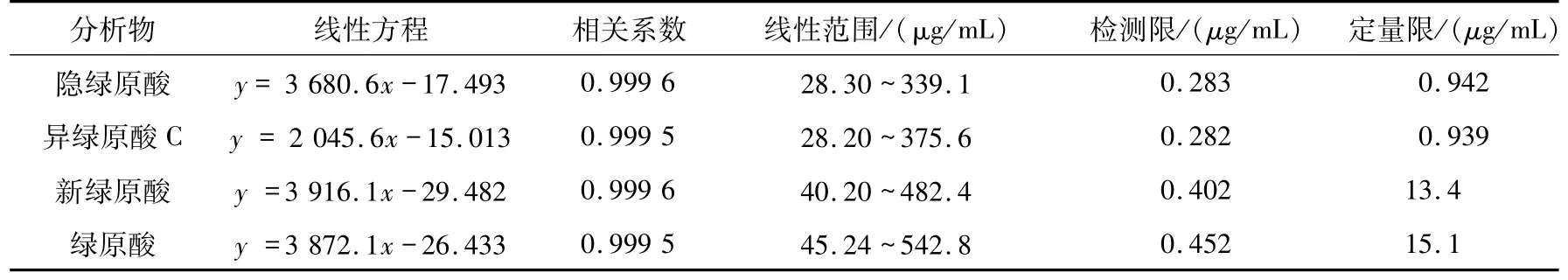
表3 4种绿原酸的线性关系Tab.3 Linear relationship of 4 chlorogenic acids
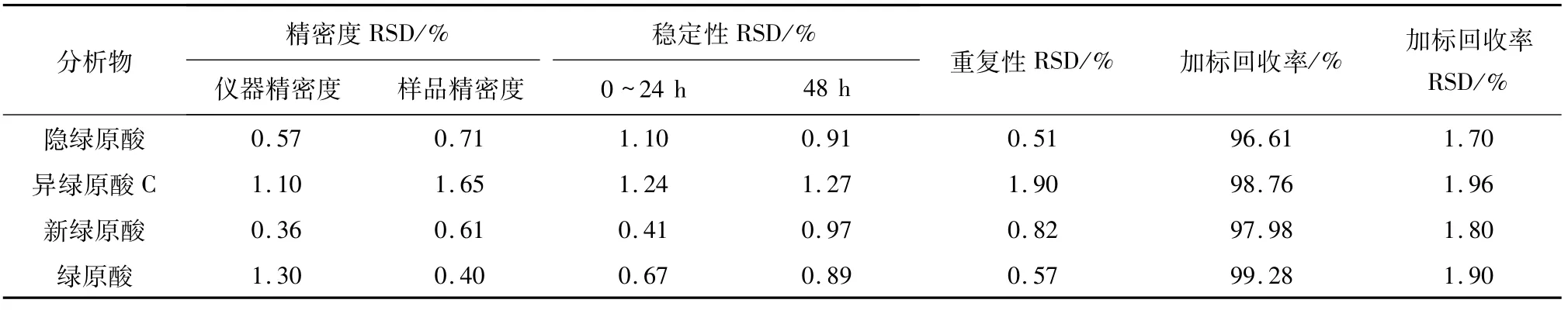
表4 精密度、重复性、稳定性和加标回收率结果(n=6)Tab.4 Results of precision,stability,repeatability and recovery(n=6)
2.3 RCF的测定及评价结果如表5所示,f1(隐绿原酸/异绿原酸C)、f2(隐绿原酸/新绿原酸)、f3(隐绿原酸/绿原酸)的RSD值分别为1.71%、1.41%、1.17%,均小于2%,说明3者的RCF都符合定量要求,以隐绿原酸为内标物来计算其余3种化合物含量的方法是切实可行的.在表6~8中的RSD值均小于2%,说明已测定的RCF值在不同的色谱柱、柱温和进样量的条件下均具有良好的稳定性.
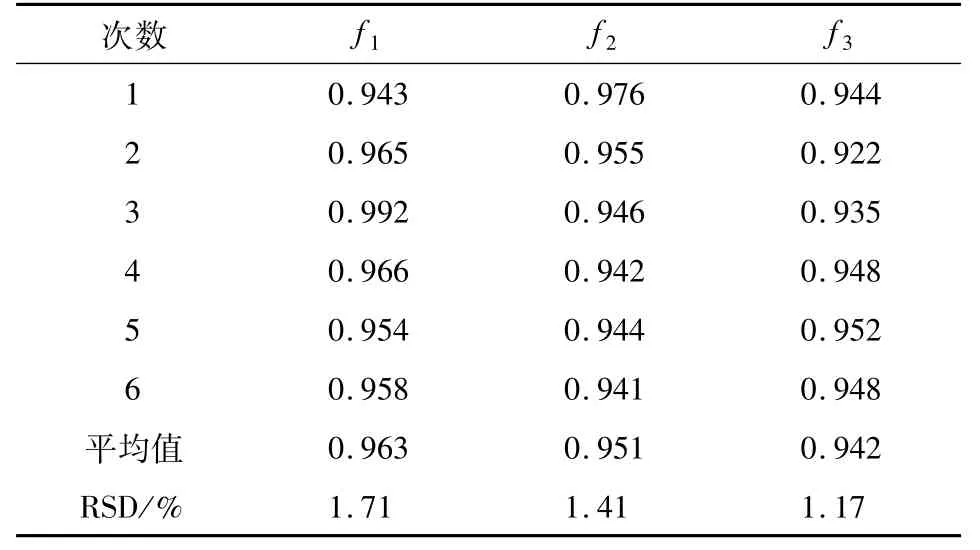
表5 相对校正因子计算结果Tab.5 Calculation results of RCFs
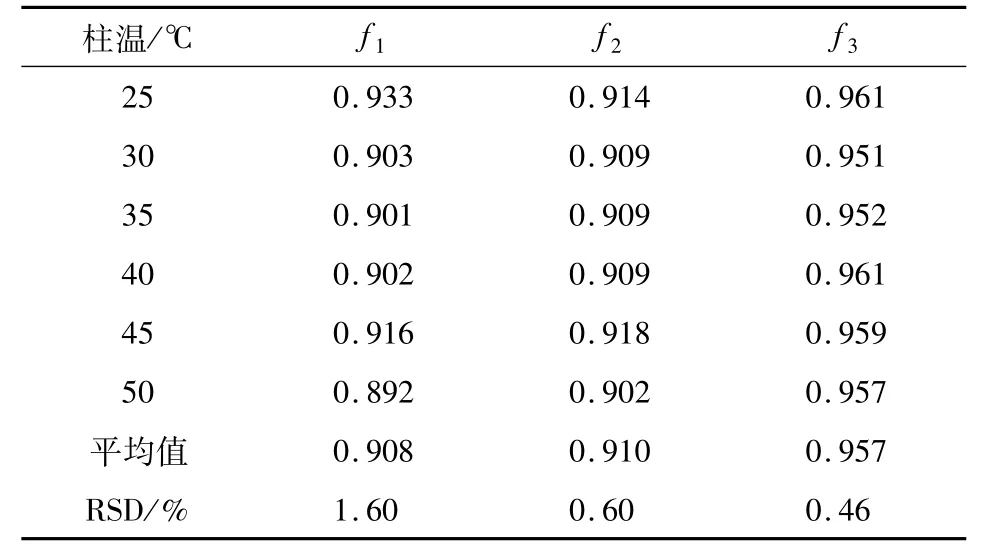
表6 不同柱温时的相对校正因子结果Tab.6 RCFs based on different column temperatures
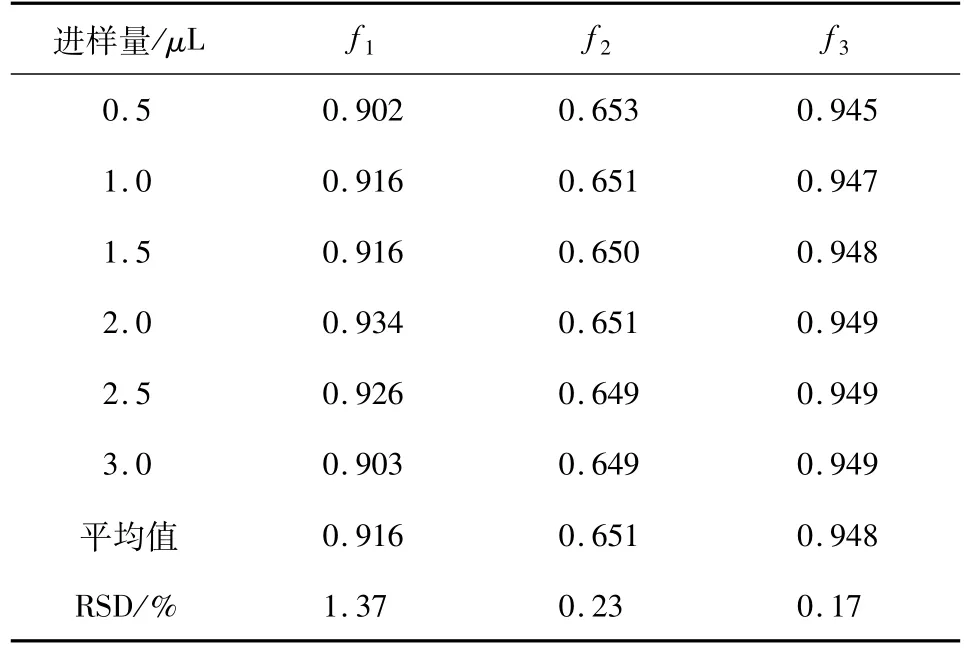
表7 不同进样量时的相对校正因子结果Tab.7 RCFs based on different injection volumes

表8 不同色谱柱时的相对校正因子结果Tab.8 RCFs based on different columns
2.4 待测成分色谱峰的定位结果如图3所示,待分析物于23 min内实现了良好的分离.异绿原酸C、新绿原酸、绿原酸的相对保留时间分别为2.015、0.626和0.908 min,其RSD值分别为1.98%、1.72%和1.85%,均小于5%,表明峰的识别结果可靠,可用于色谱峰的定位.

图3 UPLC色谱图Fig.3 Chromatograms of the sample
2.5 外标法与一测多评法的比较结果分别以一测多评法和外标法测定S1~S8样品中异绿原酸C、新绿原酸、绿原酸的含量.结果如表9所示,P值分别为0.956、0.983、0.995,均大于0.05,相关系数R均大于0.999 3,同时相对误差δ和相对标准偏差RSD值均小于5%.表明一测多评法与外标法无显著差异,说明将一测多评法应用于黑骨藤中4种绿原酸含量测定研究基本可行.

表9 外标法与一测多评法的比较结果Tab.9 Comparison of External Standard Method and QAMS
3 讨论
3.1 硅胶柱层析洗脱体系的选择由于黑骨藤中化学成分极性差距较大,为使大部分成分均能被分离出来,故本实验选取石油醚-乙酸乙酯-甲醇为洗脱体系.极性小的石油醚先除去黑骨藤中的色素、油脂等成分,再利用乙酸乙酯将中等极性成分洗脱出来,再利用V(乙酸乙酯)∶V(甲醇)=1∶1将极性大的部分洗脱出来,最后利用甲醇将硅胶柱洗脱干净,分离出不同极性片段.
3.2 一测多评法色谱条件的确定考察了流动相体系(乙腈-水、甲醇-水、乙腈-醋酸、甲醇-醋酸、乙腈-磷酸、甲醇-磷酸、乙腈-甲酸、甲醇-甲酸)和流动相pH(2.5、3、3.5、4.0),实验结果表明:当流动相为甲醇-甲酸体系且pH=3时,被检测物质峰形尖锐,无拖尾现象,且分离度都在1.5以上.运用Aglient 1290 DAD二极管阵列检测器,对隐绿原酸、异绿原酸C、新绿原酸、绿原酸进行190~400 nm全波长扫描,最大紫外吸收波长为325 nm,且分离度均大于1.5,故选择325 nm为检测波长.在这些条件下,4种绿原酸都能被较好地检测到,保障测定结果的准确性.
3.3 一测多评法内标物的确定异绿原酸C、新绿原酸和绿原酸均曾被选为内标物,然而其他3种化合物的RCF值与其RSD值的结果表明:只有当隐绿原酸作为内标物时,其他分析物RCF的RSD值才均小于2%.因此,本实验选择隐绿原酸作为内标物,利用它对另外3种成分进行定量,从而建立黑骨藤中4种绿原酸的一测多评法.
4 结论
本实验运用UPLC-Q-TOF-MS联用技术从黑骨藤提取物中共分离并鉴定出10种化合物,其中,杠柳毒 苷、periplogenin 3-O-β-D-glucopyranosyl-(1→4)-O-β-D-digitoxopyranoside和periplocymarin能诱导皮肤成纤维细胞合成胶原,与伤口愈合有关[20].新绿原酸、绿原酸、隐绿原酸能治疗免疫性关节炎,其疗效可能与抑制炎症因子有关[21].此外,原花青素A1和原花青素A2具有一定的抗过敏性,可以应用于治疗炎症和其他相关疾病[22].这种快速、高效的分析方法有利于扩展黑骨藤化学成分类别,以期进一步研究其药理活性,为黑骨藤的药效物质基础研究及质量控制提供新的依据.
本研究还建立一测多评法,以隐绿原酸为内标,同时测定异绿原酸C、新绿原酸和绿原酸的含量,并对其可行性进行了探讨分析.结果可看出,一测多评法稳定有效,能高效准确地得到其他3种分析物的含量,降低生产测试成本,实现了黑骨藤多指标成分控制的目的,而且该方法操作简便,重复性好,可为其质量评价提供参考依据.
猜你喜欢
口腔护理用品工业(2021年4期)2021-11-02
昆明医科大学学报(2021年8期)2021-08-13
天然产物研究与开发(2019年1期)2019-03-01
中成药(2018年11期)2018-11-24
天然产物研究与开发(2018年7期)2018-08-21
中成药(2018年6期)2018-07-11
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
中成药(2017年10期)2017-11-16
天然产物研究与开发(2016年11期)2016-06-15
