
基于CRISPR-Cas9 技术构建小鼠胚胎干细胞双荧光标记细胞系△
2021-09-11 05:40姜浩东匡俊企翟梓蔚赛喜雅拉图朱烁基陈寄梅
岭南心血管病杂志 2021年4期
姜浩东,匡俊企,翟梓蔚,赛喜雅拉图,朱烁基,陈寄梅,朱 平,
[1.华南理工大学医学院,广州 510006;2.中国科学院广州生物医药与健康研究院,广州 510530;3.广东省心血管病研究所广东省人民医院(广东省医学科学院),广州510080]
胚胎干细胞(embryonic stem cells,ESCs)是来自囊胚内细胞团的多能性细胞,具有自我更新和多向分化潜能,在一定条件下可以分化成体内所有细胞类型[1-2],为早期胚胎发育、再生医学、体外疾病模型构建等研究提供了良好的载体[3-5]。多种转录因子、表观遗传修饰因子、信号蛋白等在ESCs 维持更新和分化过程中发挥重要作用,独特的转录调控网络、稳定的表观遗传状态和特定的细胞周期谱也是将ESCs与高度分化的体细胞区分开来的特征[6-9]。然而,ESCs 这些特性背后的分子机制还不完全清楚。p53 是一种序列特异性转录因子和抑癌因子,对维持机体基因组稳定性至关重要[10-11]。在无应激条件下,p53 在体细胞内保持低表达水平,在多种应激压力和致癌因素介导下,p53 可通过各种转录后修饰使其表达量上调并通过直接调控多种生物学过程相关基因如细胞周期阻滞[12-13](p21)、凋亡[14](Puma、Noxa)、衰老[15](PAI-1)等来保持体细胞基因组的稳定性。ESCs在生长特性、分化能力、基因调控网络、代谢特征等方面与癌细胞具有相似处,但p53 在ESCs 中的作用目前尚不完全清楚。
Nanog是小鼠ESCs(mouse embryonic stem cells,mESCs)多能性标志之一,对维持mESCs 多能性状态十分重要[16-17]。Nanog 是mESCs 核心多能性标志中唯一在细胞间存在异质性表达的转录因子,Chambers[18]首次报道了这一现象并证实Nanog 的表达在细胞间存在异质性。此外,Chambers 还发现Nanog 低表达的细胞虽然仍可形成克隆,但是其形成克隆的数量显著低于Nanog 高表达细胞。Nanog 敲除不影响体外培养过程中mESCs 自我更新及向三胚层分化的能力,但在体内发育过程中,不能形成生殖细胞。后续有多项研究[19-23]对Nanog 异质性进行了探讨,但其背后的转录调控机制仍不清楚。
基因编辑技术是精准研究基因功能的一个重要手段,基因编辑手段经过多代发展,已由最初的锌指核酸酶(ZFNs)[24]、转录激活样效应因子核酸酶(TALEN)[25]介导的基因编辑发展至利用CRISPR-Cas9 技术[26-27]介导的基因编辑,CRISPR-Cas9技术具有制作简单、周期短、突变效率高以及成本低等特点,可根据不同策略灵活实现基因的敲除及敲入。而基于基因敲入的荧光报告基因细胞系的构建是研究基因功能的重要工具,本研究采用CRISPR-Cas9 基因敲除结合同源重组的方法构建了小鼠ESCsp53-EGFP、Nanog-mcherry 双荧光标记细胞系,并在此基础上对p53 在mESCs 中的作用进行初步探讨。
1 材料方法
1.1 实验细胞
小鼠ESCs 细胞系(129)、饲养层细胞来自中国科学院广州生物医药与健康研究院裴端卿实验室馈赠。
1.2 实验试剂与仪器
实验试剂与仪器包括N2-supplement(gibco)、B27-supplement(gibco)、非必须氨基酸(non-essential amino acids,NEAA)(gibco)、谷氨酰胺(gibco)、丙酮酸钠(gibco)、β-巯基乙醇(2-ME)(gibco)、小分子化合物PD0325901(MCE)、小分子化合物CHIR99021(MCE)、小鼠白血病抑制因子(mouse leukemia inhibitory factor,MLIF)(Millipore)、Knock-Out DMEM(gibco)、高糖DMEM(gibco)、胎牛血清(RMBI,NTC)、0.25%胰酶(gibco)、限制性内切酶(NEB)、10×NEB Buf2(NEB)、Solution I(NEB)、筛选抗生素puro、blasticidin(Thermo Scientific)、质粒小提试剂盒(天根)、lipo3000 转染试剂盒(Invitrogen)、OptiMEM(gibco)、NP40 裂解液(Thermo Scientific)、聚合酶链反应(polymerase chain reaction,PCR)试剂(Vazyme)、T7 内切酶鉴定试剂(NEB)、小鼠p53 抗体(cell signaling technoligy)、小鼠Nanog抗体(Bethyl Laboratories)、PCR 仪(BIO-GENER)、金属浴(奥盛)、感受态细胞、分光光度计(天根)、生物安全柜(Thermo Scientific)、细胞培养箱(Thermo Scientific)、流式细胞分析仪(BD LS-RFortessa X-20)、流式细胞分选仪(BD FACSAria Ⅲ)。
1.3 实验方法
1.3.1 胚胎干细胞的复苏、培养与传代 饲养层细胞培养基:10%胎牛血清(NTC)、NEAA(100×)、谷氨酰胺(100×)、高糖DMEM。转染培养基(serum+2i+lif):15%胎牛血清(RMBI)、NEAA(100×)、谷氨酰胺(100×)、丙酮酸钠(100×)、β-巯基乙醇(0.1 mmol/L)、PD0325901(2 000×)、CHIR99021(2 000×)、mLif(5 000×)、高糖DMEM。基因编辑细胞维持培养基(N2+B27+2i+lif):N2(200×)、B27(100×)、NEAA(100×)、谷氨酰胺(100×)、丙酮酸钠(100×)、β-巯基乙醇(0.1 mmol/L)、PD0325901(2 000×)、CHIR99021(2 000×)、mLif(5 000×)、高糖DMEM与Knock out DMEM 1:1 混合。细胞复苏:提前1 d 复苏饲养层细胞以合适密度种下,并用10%胎牛血清培养基培养,待饲养层细胞贴壁后,以serum+2i+lif 培养基复苏mESCs 并以合适密度种下,密度合适即传代。细胞传代:培养皿提前用0.1%明胶包板0.5 h 以上,DPBS 洗涤细胞后加入0.25%胰酶消化,离心后重悬细胞,以合适比例种下,密度合适即传代。
1.3.2 p53-sgRNA 基因敲入质粒构建,转染及T7内切酶切割鉴定 依据NCBI 基因序列在sgRNA设计网站(CHOPCHOP,http://chopchop.cbu.uib.no/)设计3 对sgRNA,选取原则参考网站排名及切割效率,引物上下游两端加入Bbs1 酶切位点,将引物退火后[退火条件:正链3 μL(100 μmol/L)、负链3 μL(100 μmol/L)、10×NEB Buffer2 5 μL、H2O 39 μL,退火程序为95℃(10 min),95℃~85℃(每秒减1℃),85℃~25℃(每秒减1℃),16℃(保存)与Bbs1 酶切后的PX330 载体连接[连接条件:Solution I 5 μL、退火片段4 μL、载体(50 ng),16 ℃2 h],提前取感受态细胞于冰上融化,向连接产物中加入30 μL 感受态细胞,冰上放置30 min,随后在超净工作台内用无菌涂布棒将其涂在含有氨苄青霉素的LB 固体培养基上,倒置放于37 ℃恒温培养箱中培养16 h,随后分装5 mL 含有氨苄青霉素的LB 液体培养基,将单个菌落挑至培养基中,37℃恒温摇床培养16 h,用质粒小提试剂盒进行质粒提取,并送测序。选择测序正确的质粒,经由脂质体转染的方法转染进入mESCs,转染体系:在24 孔板中,optiMEM(50 μL/孔)、sgRNA(2 μg/孔)与p3000(1.5 μL/孔)混合后加入lipo3000(1.5 μL/孔)与optiMEM(50 μL/孔)的混合物,静置10~15 min,悬浮转染mESCs,细胞密度为5×104/孔,转染培养基为serum+2i+lif 培养基。转染8 h 后换液,36 h 后收样并用20 μL NP40 裂解液裂解(裂解条件为56℃,99℃10 min),以裂解产物为模板,利用设计好的扩增引物进行PCR 扩增,将PCR 产物进行T7内切酶鉴定,跑胶判断条带是否正确。
1.3.3 p53-donor 载体构建,共转染,筛选及敲入鉴定 根据选择的sgRNA 切割位点,利用同源重组的原理将小鼠基因组上切割位点上下游各1 000 bp(去除转录中止位点)构建至D5S donor 载体上,经过重组、感受态细胞转化、涂板、挑菌扩增、质粒提取并经测序正确后,与sgRNA 以脂质体共转染的方式悬浮转染mESCs,转染体系为8 h 后换液,48 h 后加含嘌呤霉素的培养基(2 μg/mL)筛选,筛选后传代并挑单克隆,将单克隆一传二传代,分别用于保种与鉴定。
1.3.4 p53EGFP、Nanogmcherry 双荧光报告细胞系构建 在纯合p53EGFP/EGFP 敲入细胞系构建基础上,基于同样的原理,在Nanog 启动子后方插入红色荧光蛋白mcherry,首先根据Nanog 终止密码子附近基因组序列设计sgRNA 并构建sgRNA,经过sgRNA 切割效果验证后选定合适的sgRNA 并在此基础上设计并构建Nanog-donor 载体,donor 载体构建选用Blastcidin 作为筛选抗性,随后脂质体转染双载体进行荧光蛋白表达基因插入操作,使用Blastcidin 进行细胞筛选(饲养层细胞因其无分裂能力,故可通过多次传代将其从靶细胞中去除),最后进行纯杂合验证后得到纯合及杂合p53EGFP、Nanogmcherry 双荧光报告细胞系。
1.3.5 免疫荧光 提前制备24 孔板细胞爬片,用0.1%明胶包板并种下细胞,待合适密度后用DPBS洗涤细胞,4%多聚甲醛500 μL 固定0.5 h,DPBS 洗涤1 次,500 μL 封闭通透液作用40 min,DPBS 洗涤1 次,用封闭通透液按合适比例配置一抗(p53 1∶2 000,Nanog:1000)并在湿盒内将玻片细胞面朝下孵育1.5~2.0 h,DPBS 洗涤4 次,每次5 min,用封闭通透液配置二抗(1∶500),湿盒内避光孵育1.0~1.5 h,DPBS 洗涤5 次,每次5 min,用封闭通透液配置DAPI(1∶4 000),将玻片细胞面朝上放于原孔中,加入DAPI 后染色1 min,DPBS 洗涤3 次,每次5 min,将玻片细胞面朝下置于滴有封片剂的盖玻片上,4℃避光报存。
1.3.6 流式细胞分析 用0.25%胰酶将敲入细胞系完全消化成单细胞,用DPBS 重悬至合适密度后利用流式细胞分析的方法分析敲入细胞的绿色荧光蛋白发光强度在细胞群体内的分布情况。
1.3.7 流式细胞分选 用0.25%胰酶消化细胞后,用分选液(1 mmol/L EDTA+1%BSA)重悬细胞,根据荧光强度用流式分选仪将选定细胞分选出来。
1.4 统计学分析
2 结果
2.1 p53-EGFP knock-in 细胞系构建策略
在NCBI 上下载小鼠p53 基因组序列,将终止密码子附近100 bp 左右序列复制到sgRNA 设计网站中,综合考量切割位点及预期切割效率,切割位点距离终止密码子越近,预期切割效率最高为优选,选择三条sgRNA 作为候选sgRNA,并依次经过退火连入pX330 载体中进行初步切割效果验证。选定合适的sgRNA 并将切割位点下游1 000 bp 的基因组序列作为同源重组载体的3′arm,而将终止密码子上游1 000 bp 作为同源重组载体的5′arm,完整构建p53 基因敲入的同源重组载体。sg-RNA载体及Donor 载体如图1。
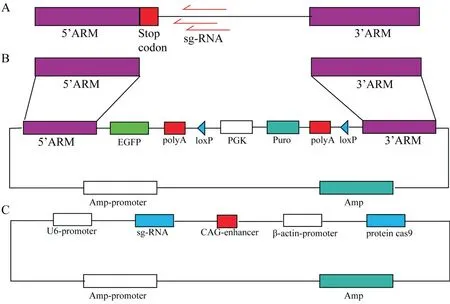
图1 p53-EGFP knock-in 细胞系构建策略图(A:sgRNA 设计位点;B:基于同源重组原理插入荧光蛋白及Donor 载体示意图;C:sgRNA 载体示意图)
2.2 p53-EGFP 敲入鉴定策略及鉴定结果
切割效率鉴定首先要利用T7 内切酶可以识别sgRNA 切割位点的特点,利用sgRNA 载体转染小鼠ESCs,经puro 筛选后利用NP40 裂解液处理后进行PCR 反应,切割效率鉴定上下游引物距离切割位点距离相差100 bp,T7 内切酶与PCR 产物作用后跑胶,选择有明显切割条带的sgRNA。挑选经过双载体共转染及puro 筛选后存活的小鼠ESCs 单克隆,并一传二分别用于保种及鉴定。使用预先设计在5′arm 同源臂、EGFP、3′arm 同源臂的鉴定引物进行p53-EGFP knock-in 细胞系纯合、杂合鉴定,若为纯合细胞,经PCR 扩增后进行琼脂糖凝胶电泳可鉴定出单个长度为1 837 bp 的条带,即鉴定引物PF与插入片段设计的引物PR-1之间的片段,未敲入细胞的条带长度为1 171 bp,即鉴定引物PF与插入片段设计的引物PR-2 之间的片段,而杂合细胞则同时出现上述两个条带(图2)。
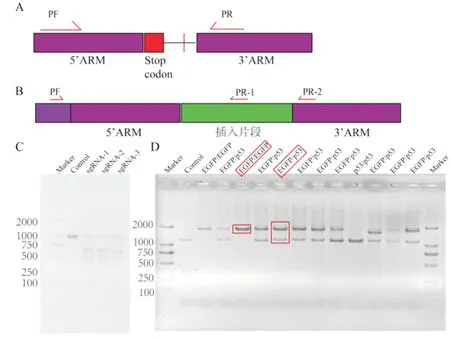
图2 p53-EGFP knock-in 细胞系敲入鉴定及纯杂合鉴定图(A:sg-RNA 切割效果验证策略;B:纯杂合敲入验证策略;C:sg-RNA 切割效果验证结果;D:纯杂合敲入验证结果)
2.3 小鼠胚胎干细胞中p53 表达具有异质性且与Nanog 表达互斥
经过鉴定得到p53-EGFP 纯合和杂合细胞系,倒置荧光显微镜下可以看到纯合、杂合细胞系与野生型对照细胞相比,有一定程度的绿色荧光出现。流式细胞分析可以看出,纯合细胞系FITC荧光峰值高于对照,而杂合细胞系则位于两者之间。免疫荧光染色发现,以免疫荧光染色的p53 的红色荧光为对照,可发现p53-EGFP 细胞系的绿色荧光较好地反映了p53表达情况,且细胞群体间p53的表达具有异质性的特点。此外,利用免疫荧光对小鼠ESCs多能性标志Nanog 进行共染后发现,p53 表达与Nanog表达呈负相关关系,即p53 高表达的细胞里Nanog表达较低,而p53低表达的细胞里Nanog表达较高。
2.4 p53 抑制小鼠胚胎干细胞克隆形成
将敲入细胞系按荧光强弱即p53 表达高低进行分选,分选比例为荧光强弱大约各20%,分选完成后再次进行流式分析,可发现纯合、杂合细胞p53 高表达细胞群体与低表达细胞群体相比为两个不同细胞群体。将分选后的细胞重新种下进行克隆形成实验,可发现,p53 高表达细胞与低表达细胞相比,克隆形成数量下降,重新消化种下一代后,可得到类似结果。
2.5 p53-EGFP、Nanog-mcherry 双荧光标记细胞系构建
基于同样的原理,在p53-EGFP knock-in 细胞系的基础上,在Nanog 终止密码子后方插入红色荧光蛋白mcherry 用于指示Nanog 蛋白的表达水平。敲除效率验证显示预先设计的三条sgRNA 均有一定的切割效果。双载体转染、筛选后,经纯合、杂合鉴定可得到Nanog-mcherry knock in 纯合及杂合细胞系。倒置荧光显微镜下可以看到纯合、杂合细胞系相比对照而言都有一定程度的红色荧光出现,将纯合敲入细胞进行固定、通透、DAPI 染色后在共聚焦显微镜下观察可发现绿色荧光与红色荧光呈互斥关系,见图3、4。
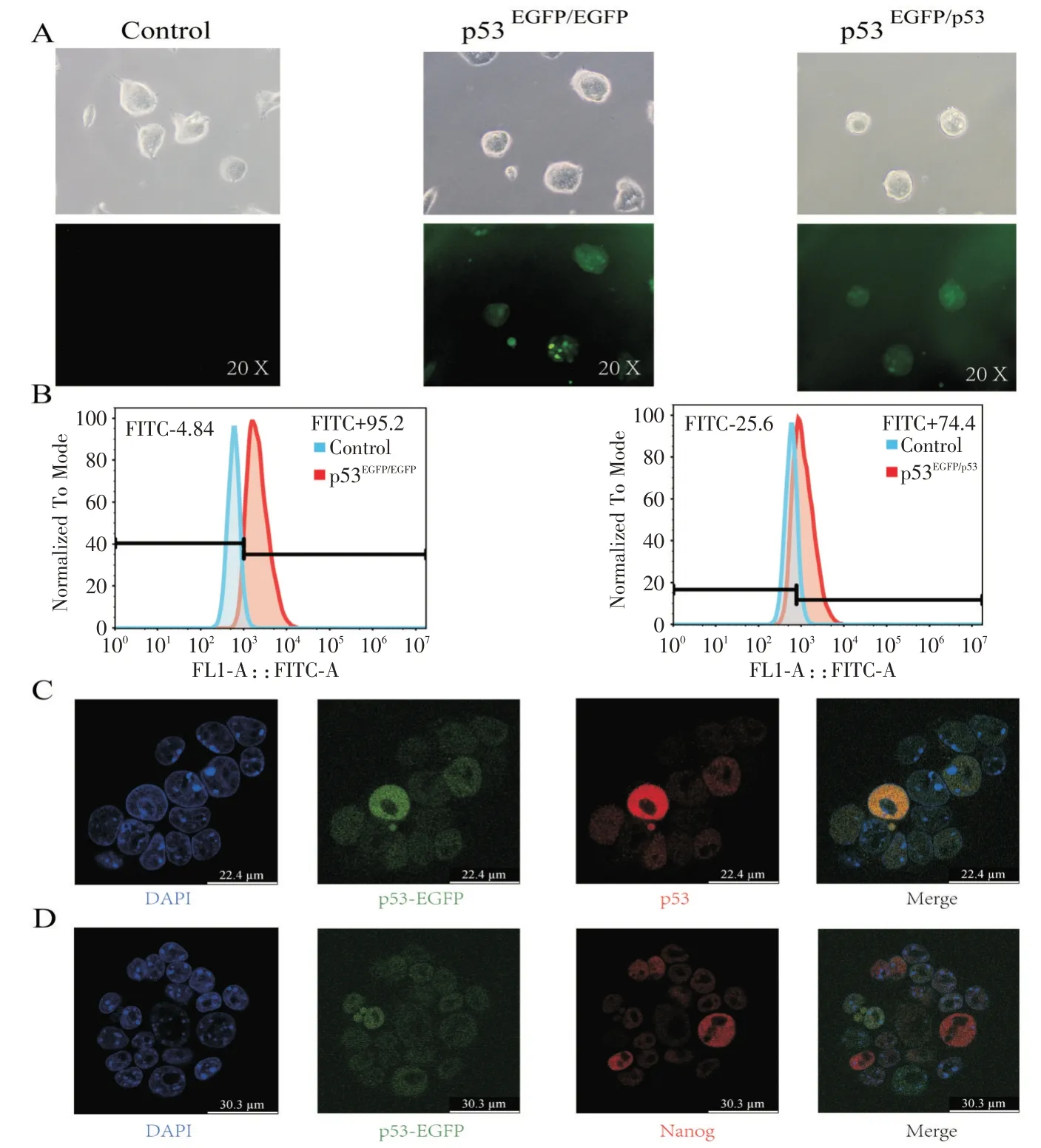
图3 小鼠ESCs 中p53 表达具有异质性且与Nanog 表达互斥(A:荧光显微镜观察对照、纯合、杂合p53-EGFP 敲入细胞系;B:流式细胞分析对照、纯合、杂合p53-EGFP 敲入细胞系荧光分布;C:p53-EGFP 绿色自发荧光与免疫荧光染色p53 红色荧光对应关系;D:p53 表达与Nanog 表达互斥)
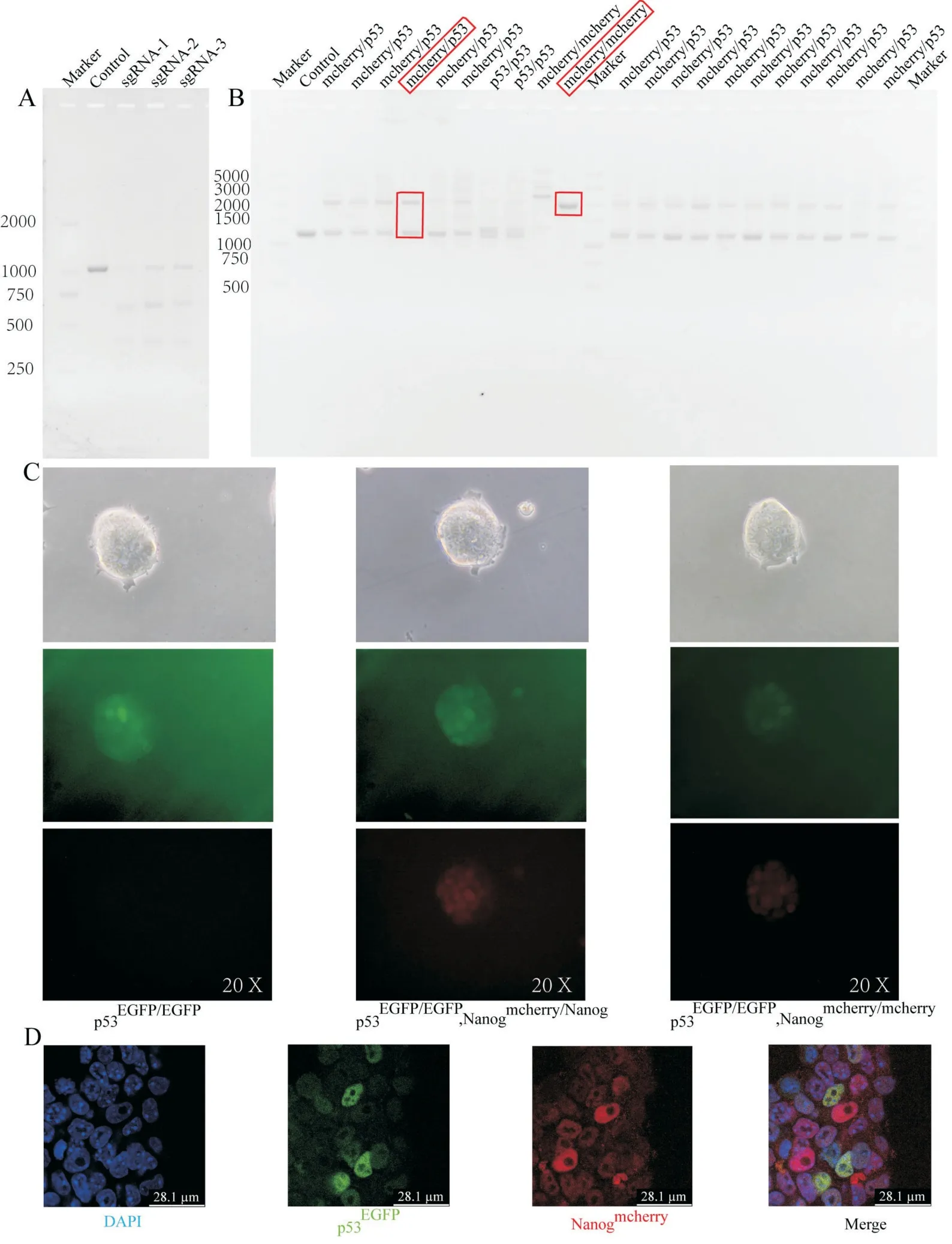
图4 p53-EGFP、Nanog-mcherry 双荧光标记细胞系构建(A:sg-RNA 切割效果验证;B:纯合、杂合敲入验证;C:倒置荧光显微镜下观察对照、纯合、杂合双荧光标记细胞;D:共聚焦观察双荧光标记细胞中p53与Nanog 互斥关系)
2.6 小鼠ESCs 中p53 可以抑制Nanog 表达
经过流式分析红色荧光表达可以发现,与对照相比,纯合Nanog-mcherry knock in 细胞系红色荧光分布峰图明显右移,而杂合敲入细胞峰图则位于纯合及对照之间,见图5。过表达p53 后可以发现红色荧光分布明显左移,表明p53 可以在Nanog 上游发挥作用,即p53可以抑制Nanog表达,见图6。

图5 p53 抑制小鼠ESCs 克隆形成(A:流式分选不同荧光强度的纯合、杂合p53-EGFP 敲入细胞;B:p53 高表达抑制小鼠ESCs 克隆形成)

图6 小鼠ESCs 中p53 可以抑制Nanog 表达(A-B:流式细胞分析对照、纯合、杂合Nanog-mcherry 敲入细胞系荧光分布;C:在纯合细胞系中过表达p53 后,红色荧光减弱)
3 讨论
CRISPR-Cas9 基因编辑工具为研究基因功能提供极大的便利,基于同源重组原理的基因敲入技术可实现在基因组特定位点特异性插入外源片段。基于此原理,本研究利用CRISPR-Cas9 基因敲入技术成功构建了小鼠ESCsp53、Nanog 双荧光报告细胞系,通过在p53 基因编码序列末端引入绿色荧光蛋白EGFP,Nanog 基因组终止密码子处引入红色荧光蛋白mcherry,理论上,p53 蛋白表达量与EGFP 相同,而Nanog 蛋白表达量与mcherry相同,因此,在荧光显微镜下分别观察EGFP、mcherry 的荧光强度即可反映活细胞状态下p53、Nanog 蛋白的表达水平,无需额外检测,为研究p53 在ESCs 命运调控中的具体作用机制提供了有力工具。
ESCs 一个重要的特性是在保持自我更新能力的同时具有向三胚层分化的能力,ESCs 如何在自我更新和分化能力之间取得平衡是干细胞相关研究中的热点问题。而细胞间异质性则可能是干细胞在对诱导分化信号做出反应的同时保持其自我更新潜力的内在机制。多项研究已报道多个与多能性相关的转录因子在ESCs 中以异质性的方式表达,如Nanog、Gata6、Rex1 等。大约10%~20%的ESCs 中不表达Nanog,Nanog 高表达细胞群体未分化克隆形成数量高于Nanog 低表达细胞,而Nanog 阴性细胞则显示出更高的分化倾向,且这种异质性的状态是处于动态波动的,即Nanog 高表达细胞与低表达细胞可以相互转化,提示ESCs 能够在偏向自我更新和偏向分化的状态之间切换,以在响应不同胞外信号时做出不同反应。
p53 作为抑癌因子在抑制成熟体细胞向无限增殖的恶性肿瘤细胞的转变过程中发挥重要作用,但在ESCs 中发挥什么样的作用目前尚未可知。有研究发现,在紫外线或Doxorubicin 的作用下,mESCs 中p53 蛋白第315 位丝氨酸可发生磷酸化进而提高其蛋白浓度,而Nanog 启动子上游存在两个p53 蛋白的结合位点,在损伤诱导条件下,p53 通过第315 位丝氨酸的磷酸化提高其蛋白浓度并招募共抑制因子mSin3a,实现对Nanog 的直接转录抑制,进而对ESCs 的细胞命运进行调控[28]。然而,在目前的相关研究中,对P53 在ESCs中作用的研究通常是在体外人为创造诱导分化环境(去除LIF)[29]或诱导内源活性氧增多(去除β-巯基乙醇)[30]等条件下得到的相应结论,在mESCs正常体外培养过程中,p53 能否在mESCs 维持自我更新中发挥一定作用以及与Nanog 是否仍具有调控关系,由于缺乏合适的细胞模型,目前尚未有相关结论。在本研究中,我们构建的P53、Nanog 双荧光标记细胞系为当前研究提供了便利,并在此基础上发现P53 在mESCs 中存在异质性表达的特点,且其异质性表达与Nanog 蛋白表达呈互斥关系,进一步发现P53 可抑制Nanog 表达,即P53 可通过调控Nanog 表达参与小鼠ESCs 正常维持培养过程中不同状态的调控,为P53 参与调控细胞命运的机制提供新的见解。
猜你喜欢
临床肝胆病杂志(2022年8期)2022-11-23
社会科学战线(2022年5期)2022-07-23
科学与生活(2021年16期)2021-11-25
西南医科大学学报(2021年5期)2021-09-23
现代企业(2021年2期)2021-07-20
种子(2021年3期)2021-04-12
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
医学研究杂志(2015年9期)2015-07-01
