
Mechanism of tumor synthetic lethal-related targets
2021-09-10 07:30:34YuhangZhangPengXu
Yuhang Zhang ,Peng Xu
1 Shaanxi University of Traditional Chinese Medicine,Xianyang 712000,China
2 Shaanxi Academy of Traditional Chinese Medicine,Shaanxi Provincial Hospital of Traditional Chinese Medicine,Xi’an 710003,China
Abstract Synthetic lethality is becoming more and more important in the precise treatment of oncology.Malignant tumors caused by gene mutations involve a complex DNA signaling process,and inhibition of DNA signaling in different ways may more effectively control the occurrence and development of tumors. Inhibition of tumor paired lethal genes effectively kills tumor cells,and more and more novel drugs that inhibit tumors are developing in this direction.This article reviews the synthetic lethal theory and discusses selection of drugs to target mutated genes in common solid tumors.The synthetic lethal gene pairs,representative targeted drugs,and related characteristics of four tumor types:lung cancer,breast cancer,colon cancer and prostate cancer,are systematically reviewed.
Key words: synthetic lethal;targeted drug;common tumors
Tumors are a distant effect produced by DNA damage in the body.Inhibition of DNA damage repair in tumor cells has a fatal impact on their development.The principle that tumor cells undergo lethal effects under two or more non-lethal and non-allelic mutations is known as a synthetic lethal.Cancer therapy has gradually developed from interfering with DNA physicochemical synthesis to precise targeting at the genetic level.With the wide application in clinical practice of gene sequencing in patients with mutant carcinogenic tumors,the application of synthetic lethal-related paired gene targeted drugs can more effectively block the proliferation of tumors and improve prospects for cancer therapy.In this review,based on the theory of tumor gene synthesis and lethality,the principle of tumor inhibition by paired genes that initiate related tumorigenesis is summarized,and the drug treatment options for common solid tumors are explored.
Synthetic lethal
Organisms have complex DNA repair systems to protect their integrity,mainly through homologous recombination pathways or non-homologous end joining,to repair double-stranded DNA breaks,and through base excision repair or mismatch repair to repair singlestranded DNA breaks.DNA damage repair is initiated after normal cellular DNA is damaged under the influence of physicochemical,biological,and other carcinogenic factors,and damage repair mechanisms are meticulous and complete to protect it from carcinogenesis.
However,tumor cells themselves have genetic defects;hence,the failure to repair DNA damage leads to fatal effects in cells.Driver mutations are an important factor in thousands of mutations leading to tumorigenesis and have a greater impact in the development of tumors than other passenger genes[1].Generally,tumors have multiple oncogene mutations,but most tumors are sensitive to the inhibition of a single oncogene,which is known as“oncogene addiction”[2].
The key goal of cancer drugs is to selectively initiate the genetic damage of target and persistent tumor cells.Under the theory of synthetic lethality,the multiple inhibition of enzymes involved in DNA repair and cell cycle control in tumor genetic signaling pathways can block the development of tumors.Synthetic lethality is important in targeted and precise treatment of tumors after chemotherapy[3–4](Fig.1).
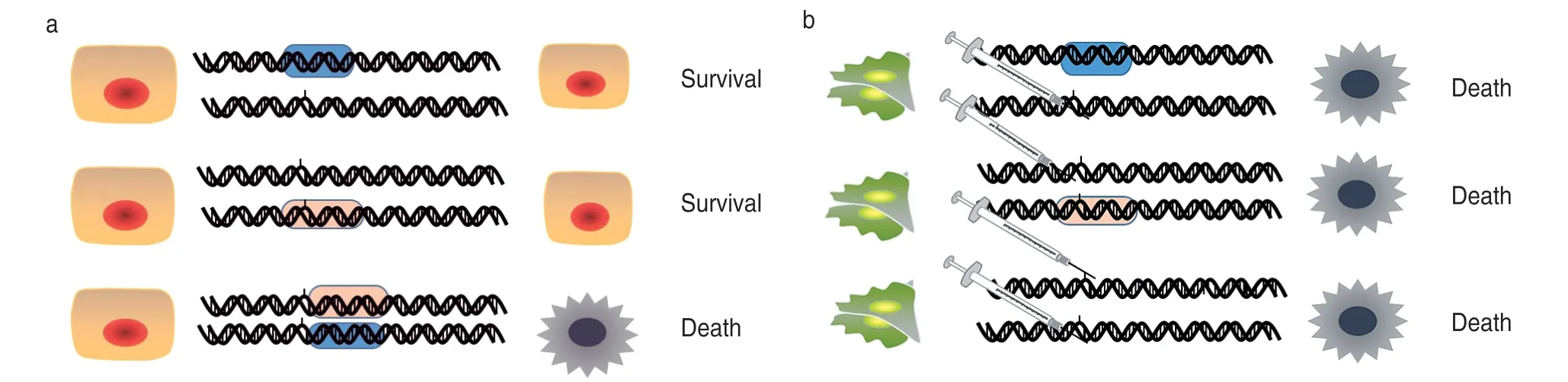
Fig.1 Cells in synthetic lethal theory
This inhibition leads to tumor cell apoptosis due to oncogene signaling addiction,collateral vulnerability,and more generally synthetic lethality[5–6].An attractive strategy for the synthetic lethal treatment of malignant tumors is to target enzyme functions[7]and related proteins that are dispensable in normal cells.This makes gene inhibition methods feasible by focusing on enzymes that are essential for the survival of mutant protocancerous cells,such as tyrosine kinases and cell cycle-dependent methionine kinases.Although many drugs that block driver gene addiction have been shown to be successful in the treatment of cancer,multiple inhibition of cancer gene mutations is still challenging in cancer therapy.For example,malignant cells generated by BRCA gene mutations are clinically effective in demonstrating the synthetic lethality of PARP (poly ADP-ribose polymerase)inhibitors[8].This example encourages modern cancer research to search for synthetic lethal“gene-drug”combinations[9]to target other mutated driver genes in the progression of cancer,toward“gene-gene”patterns.
Tumor synthetic lethal gene mechanism
Lung cancer
Lung cancer is still the most common tumor worldwide.Since the correlation between the occurrence of lung cancer and driver genes has been increasingly confirmed,the application of targeted drugs in the“genedrug”model of precise treatment of lung cancer has been widely and meaningfully effective.
EGFR-KRAS
Epidermal growth factor (EGF) converts extracellular signals into appropriate cellular responses via epidermal growth factor receptor (EGFR) tyrosine kinase binding ligands and activating multiple signaling steps[10].EGFR’s known ligands include EGF,TGFA/TGF-α,AREG,epigen/EPGN,BTC/betacellulin,HURP/EREG and HBEGF[11].Ligand binding activates at least four major downstream signaling cascades,including RAS-RAFMEK-ERK,PI3K-AKT,among others.EGFR may also activate the NF-κB signaling cascade[12]and can also directly phosphorylate other proteins such as RGS16,activating its GTPase activity and may transduce EGF receptor signal to G protein-coupled[13]receptor signal transduction[14].It phosphorylates MUC1 and increases its interaction with SRC and CTNNB1/β-catenin.Cell migration is positively regulated by interaction with CCDC88A/GIV,which retains EGFR at the cell membrane after ligand stimulation,thereby promoting EGFR signaling and thus triggering cell migration[15].The KRAS gene belongs to the RAS gene family,in which the Ras protein binds GDP/GTP and has intrinsic GTPase activity.Its related pathways include the common cytokine receptor gamma chain family signaling pathway and the negative regulation of the MAPK pathway.Gene Ontology (GO) annotations related to this gene include GTP binding.An important paralog of this gene is NRAS,which plays an important role in the regulation of cell proliferation[16]and in promoting oncogenic events by inducing transcriptional silencing of tumor suppressor genes (TSG) in lung cancer cells in a ZNF304-dependent manner[17].KRAS and EGFR proteins produce synthetic lethality when co-expressed in human lung adenocarcinoma (LUAD) cells[18].The clinical use of KRAS-associated inhibitory drugs in combination with EGFR-TKIs is increasingly widely recommended in the progression of lung cancer treatment (Fig.2a).
CDK4-RAS
Serine/threonine protein kinase,also known as cell cycle regulatory protein (CDK),is involved in the control of the cell cycle and differentiation,promoting G1/S transition.Phosphorylated pRB/RB1 and NPM1.CDK interacts with D-type G1 cyclins during the interphase of G1 to form the pRB/RB1 kinase and to control cell cycle entry and is also involved in the initiation and maintenance of cell cycle exit during cell differentiation.CDK prevents cell proliferation and negatively regulates cell differentiation and may act in centrosome organization and delay senescence during the cell cycle stage[19].It plays an important role in cellular regulation,and abnormal expression of this gene in tumor cells leads to continuous malignant proliferation of tumors.CDK1 is a target of miR-34c-3p,which is one of the promising lethal targets for KRAS mutant cancers.In addition,the combination of CDK1 inhibition (mediated by RO3306)and miR-34c-3p overexpression leads to an additive effect on the viability of cells expressing KRASmut[20].KRAS is involved in transcriptional activation and repression of selected genes through chromatin remodeling(alterations in DNA-nucleosome topology).Components of the SWI/SNF chromatin remodeling complex perform key enzymatic activities to alter chromatin structure by altering DNA-histone contacts in the nucleosome in an ATP-dependent manner.The recruitment of CREBBP to the promoter is increased by a CREST-dependent mechanism,which leads to transcriptional activation.During cell differentiation,the role of KRAS and CDK4 in regulating cell proliferation is very important,and the addition of CDK cell cycle inhibitors to KRAS monophasic blockade can effectively arrest the growth of cancer cells[21](Fig.2b).

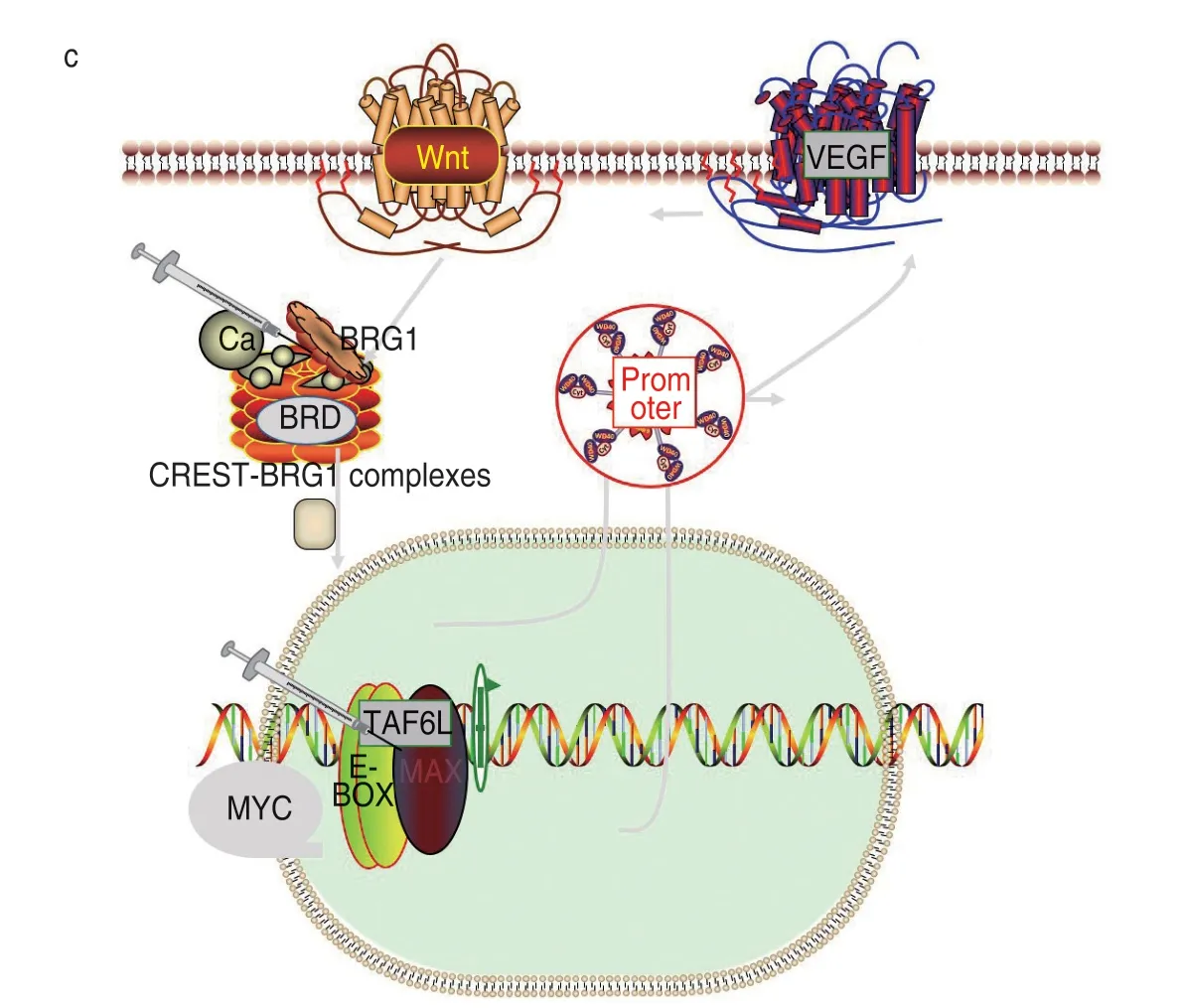

Fig.2 Synthetic lethal target pair gene signal pattern diagram
BRG1-MYC
BRG1 is a downstream protein of SMARCA4 (actindependent modulator),and its related pathways include Wnt-mediated regulation of β-catenin signaling and transcription of target genes.The downstream component complex of CREST-BRG1 associated with BRG1,a multiprotein complex,regulates promoter activation by coordinating the release of calcium-dependent repressor complexes and the recruitment of activator complexes,and inhibition of BRG1 protein inhibits the transcriptional priming program of the genetic material[21].Its protein bromodomain (BRD) is an epigenetic reader domain that selectively recognizes acetylated lysine residues on histone protein tails and is the only known protein module that can target acetylated lysine residues.The MYC gene is closely related to the inhibition of cell differentiation and tumor transformation.The protein encoded by this gene forms a heterodimer with the related transcription factor MAX[22].This complex binds to the EboxDNA consensus sequence and regulates transcription of specific target genes together with TAF6L to activate target gene expression by RNA polymerase II pause release (by similarity).Modulators involved in somatic cell reprogramming control the self-renewal of embryonic stem cells[23].It activates the transcription of genes associated with growth.It binds to the VEGFA promoter and promotes the production of VEGFA and subsequent germination of angiogenesis[24].It is involved in tumor-associated angiogenesis.There is an antagonistic functional link between BRG1 and MYC and,therefore,non-compliance to RA and GC via BRG1 inactivation involves deregulation of MYC activity.Mechanistically,some of these roles are mediated by binding of BRG1 to MYC and MYC target promoters[25].BRG1-MYC cotargets inhibition from the promoter to lead to tumor cell suppression[26](Fig.2c).
TMPRSS4-DDR1
Transmembrane serine protease[27](TMPRSS4)encodes a protein that binds together with an N-terminal anchor sequence and a glycosylated extracellular region containing a serine protease domain.Discoidin domain receptor tyrosine kinase (DDR) encodes a protein belonging to the subfamily of tyrosine kinase receptors,which regulates cell attachment to the extracellular matrix,remodeling of the extracellular matrix,cell migration,differentiation,and cell proliferation,and plays an important role in tumor cell invasion.There is a consistent co-expression between TMPRSS4 and DDR1[28].Like TMPRSS4,the DDR1[29]promoter is hypomethylated in NSCLC,while hypomethylation is an independent prognostic factor for disease-free survival.Treatment with 5-azacytidine increased DDR1 levels in the cell lines,indicating epigenetic regulation.Cells lacking TMPRSS4 are highly sensitive to the cytotoxic effects of dasatinib,a DDR1 inhibitor[30].TMPRSS4/DDR1 double knockout (KD) units,but none of the KD cells suffered G0/G1 cell cycle arrest,loss of E2F1 and cyclins A and B,elevated p21 levels as well as massive apoptosis.Studies have shown in vivo tumor regression in mice injected with double KD.A synthetic lethal interaction between DDR1 and TMPRSS4 has been identified resulting in a new vulnerability in NSCLC (Fig.2d).
Breast cancer and ovarian cancer
Breast cancers have gradually become the malignant tumors with the highest incidence in the world in recent years,which is still a complex link in pathogenesis,but gene mutation is still an important factor in tumorigenesis,with the increase of the affected population and the extension of survival.The evidence for mutation continuation and accumulation through offspring is increasing,and inhibition of breast and ovarian cancer is an example of synthesis leading to persuasive and clinical validation.
BRCA-PARP
Most breast and ovarian cancers of BRCA-PARP are accompanied by BRCA gene mutation[31].The BRCA1/2 gene,as a common tumor suppressor gene,accounts for about 40% of hereditary breast cancers and more than 80% of hereditary breast and ovarian cancers.BRCA1/2 mainly encodes DNA damage repair-related proteins as well as other tumor suppressors,which combine with DNA damage sensors and signal transducers to form a large multi-subunit protein complex[32],and also mediates the control of R-loop-related genomic instability involved in double-strand break repair and/or homologous recombination[33].Members of the PARP gene (poly (ADP-ribose) polymerase) family (PARP1,PARP2,PARP3,etc.) are common nuclear proteins with similar main roles in gene function and are involved in the regulation of various important cellular processes[34].The BRCA gene requires phosphorylation of the encoded substrate for normal cell cycle progression from G2 to mitosis upon posttranslational modification.PARP1-dependent PARP9-DTX3L-mediated ubiquitination[35]can promote the rapid and specific recruitment of BRCA1 to sites of DNA damage[36].In the case of BRCA1 mutations affecting BARD1 heteromerization,PARP inhibitors will reduce PAR formation and the rapid recruitment to DNA damage sites by the BRCA1/BARD1 complex,thereby inhibiting HR-mediated repair and inducing cell death.The current combination of breast and ovarian cancer patients with germline lesions can effectively inhibit breast and ovarian cancer[37–39](Fig.3).
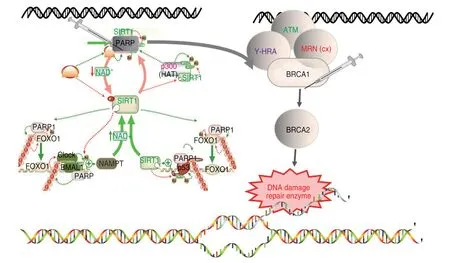
Fig.3 Synthetic lethal target pair gene signal pattern diagram
Colon cancer
An important point in the tendency of colon cancer to be younger may be related to its continuous repair defect of gene instability.Normal mismatch repair plays an extremely important role in body cells,and the loss of this function leads to complex changes in other signaling activities of cells.Inhibition of colon cancer tumor cells requires multiple pathways.
PLK1-RAS
The PLK1 gene belongs to the CDC5/Polo subfamily,and highly conserved structural protein kinases are closely related to cell cycle progression,mitosis,and DNA damage.It is highly expressed during mitosis and performs several important functions throughout the M phase of the cell cycle[40],including regulation of centrosome maturation and spindle assembly,inhibition of anaphase-promoting complex/ring inactivation(APC/C),and regulation of mitotic exit and cytokinesis by phosphorylating adhesin subunits (e.g.,STAG2/SA2)to regulate adhesin dissociation from chromosomes.Phosphorylated SGO1:SGO1 isomer 3 is required for spindle pole localization and plays a role in regulating its centriolar cohesion function[41].It mediates the phosphorylation of FBXO5/EMI1 (a negative modulator of the APC/C complex) during prophase,leading to FBXO5/EMI1 ubiquitination and degradation by the proteasome.It also acts as a negative modulator of p53 family members and phosphorylates TOPORS,thereby inhibiting the sulfonylation of p53/TP53 and simultaneously enhancing the ubiquitination and subsequent degradation of p53/TP53.It phosphorylates the transactivation domain of the transcription factor p53/TP53,thereby inhibiting p53/TP53-mediated transcriptional activation and proapoptotic functions[42].Combined blockade of PLK1-KRAS effectively inhibits colon cancer[43].However,relevant studies on clinical application are still in progress(Fig.4a).
PLK1-TP53
PLK1 is highly expressed during mitosis,and TP53,when associated with the CAK complex in response to DNA damage,prevents CDK7 kinase activity and thus cell cycle progression.Isoform 2 enhances the transactivation activity of isoform 1 of some,but not all,TP53-inducible promoters[44].Isotype 4 inhibits transactivation activity and weakens growth inhibition mediated by isotype 1.Isotype 7 inhibits isotype 1-mediated apoptosis[45].TP53TG1 can regulate PLK1.Yang Ping[46]could induce OCI-ly1 and OCI-ly3 cells to arrest in G2 phase and reduce cell migration and invasion by up-regulating lncRNATP53TG1 in OCI-ly1 cells and down-regulating lncRNATP53TG1 in OCI-ly3 cells,and the mechanism may be through the regulation of PLK1.Application of PLK1 inhibitors blocks the signaling of TP53 and inhibits tumorigenesis[47](Fig.4b).
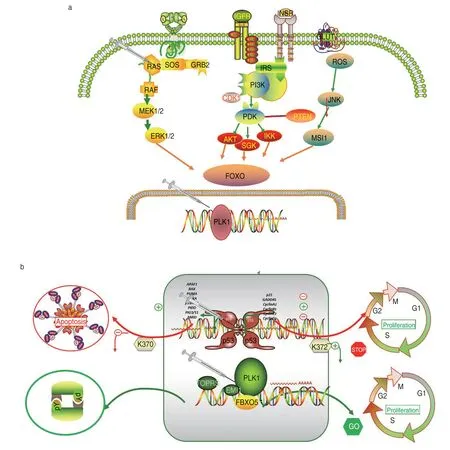
Fig.4 Synthetic lethal target pair gene signal pattern diagram
Prostate cancer
Prostate cancer plays an important role in new tumors in men,and its related research progress is becoming more and more extensive.The relevant combinations currently inhibiting prostate cancer with a synthetic lethal strategy are as follows.
BCL2-PTEN
BCL2 is a protein-coding gene.Its encoded protein regulates cell death by controlling mitochondrial membrane permeability.Its associated pathways include apoptosis regulation as well as T cell and Nur77 signaling.PTEN antagonizes the PI3K-AKT/PKB signaling pathway by dephosphorylating phosphoinositides[48],thereby regulating cell cycle progression and cell survival[49].The unphosphorylated form cooperates with AIP1 to inhibit AKT1 activation.It dephosphorylates tyrosine-phosphorylated focal adhesion kinase and inhibits cell migration as well as integrin-mediated cell spreading and focal adhesion formation,acting as a key modulator of the AKT-mTOR signaling pathway.Among several genetic alterations involved in prostate cancer development,BCL2 is an important target molecule after ablation or castration of androgen-independent prostate cancer (AIPC)[50].BCL-2,implicated in proandrogenrelated signaling during the progression of androgenindependent prostate cancer (ADPC),is a survival molecule,while BCL-2 upregulation,PTEN loss,PI3K/AKT phosphorylation and receptor tyrosine kinase (RTK)activation are mainly associated with AIPC.Guccini[51]showed that PTEN deficiency or chemotherapy-driven aging limited the progression of prostate cancer in mice.The nucleo-monoubiquitinated[52]form of PTEN has apoptotic potential,while the cytoplasmic nonubiquitinated form induces weaker tumor inhibition (Fig.5a).
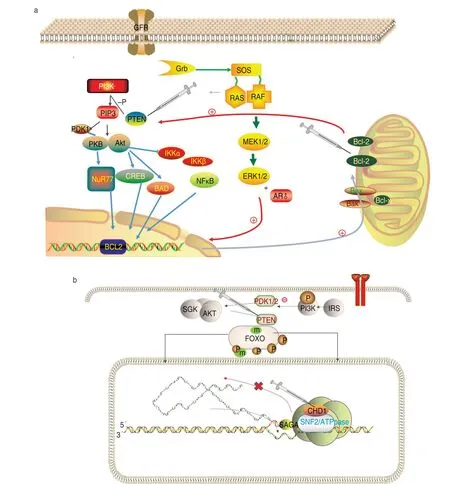
Fig.5 Synthetic lethal target pair gene signal pattern diagram
CHD1-PTEN
Loss of the CHD1-PTEN gene encoding the chromatin remodeler CHD1 is the most common alteration in prostate cancer.The CHD protein family is characterized by a chromatin domain and a SNF2[53]-associated helicase/ATPase domain.CHD genes may alter gene expression by modifying chromatin structure,thereby negatively regulating the transcription of their chromosomal DNA templates.Augello[54]demonstrated that CHD1 occupies a prostate-specific enhancer and is rich in androgen receptor (AR) and lineage-specific cofactors.Prostate tumors with loss of CHD1 appear to be highly sensitive to abiraterone treatment[55].CHD1ATP-dependent chromatin remodelers regulate the transcription of polymerase II.They can act as a substrate recognition component of the transcriptional regulatory histone acetylation (HAT) complex SAGA[56].Efficient transcription by RNA polymerase I,and more specifically,the polymerase I transcription termination step,is also required to negatively regulate DNA replication.CHD1 is not only involved in transcription-associated chromatin remodeling,but also requires the maintenance of specific chromatin configurations throughout the genome.CHD1 is also associated (by similarity) with histone deacetylase(HDAC) activity.CHD1 is required to bridge the SNF2,FACT complex,PAF complex,and the U2snRNP complex to H3K4me3.CHD1 regulates the function of pre-mRNA splicing efficiency by the physical bridging of some spliceosome components to H3K4me3[57].Maintenance of open chromatin and pluripotency in embryonic stem cells is required through similarity.CHD1 is characterized by synthetic lethality with PTEN inhibition of prostate cancer[58](Fig.5b).
Other tumors and genes
CHK1
The protein encoded by CHK1,CHK1 gene belongs to the Ser/Thr protein kinase family.Checkpoint-mediated cell cycle arrest is required in response to DNA damage or the presence of unreplicated DNA.The role of this protein is to integrate signals from ATM and ATR,two cyclins involved in the DNA damage response,which are also associated with chromatin in meiotic prophase I.Phosphorylation of CDC25A protein phosphatase by this protein is a double-stranded DNA break necessary for cells to delay cell cycle progression in response.
Its related pathways include p53 signaling and DNA damage ATM/ATRG1/S checkpoint,regulating the phosphorylation of CDC25A at“Ser-178”and“Thr-507”and the phosphorylation of CDC25C at“Ser-216”resulting in binding sites for 14-3-3 proteins,which inhibit CDC25A and CDC25C.The newly synthesized bis (indolyl) thiazole alkaloid analogue,nortopsentin234(NORA234),resulted in an initial decrease in the proliferation and clonogenic potential of CRC spherical cells (CR-CSphCs),followed by an adaptive response to select CR-CSphC resistance compartments.Cells that were spared by treatment with NORA234 expressed high levels of CD44v6,a synthetic lethal mode caused by constitutive activation of the Wnt pathway.
The Rad51-WEE1
WEE1 gene encodes a nuclear protein,which is a tyrosine kinase belonging to the Ser/Thr family of protein kinases.This protein catalyzes the inhibitory tyrosine phosphorylation of CDC2/cyclin B kinase,and appears to coordinate the transition between DNA replication and mitosis by protecting the nucleus from cytoplasmically activated CDC2 kinase.For Wee1 kinase and Rad51 recombinase in head and neck tumors are two proteins involved in regulating replication stress and homologous recombination repair in cancer cells.
Synergism between Rad51 inhibitor (B02) and Wee1 inhibitor (AZD1775) is associated with forced CDK1 activation and decreased Chk1 phosphorylation,leading to excessive DNA damage and replication stress,ultimately leading to abnormal mitosis and apoptosis.
NRN1
NRN1 protein contains a consensus cleavage signal protein found in glycosylphosphatidylinositol (GPI)-anchored protein quality.It promotes neurite growth and arborization and has a role in promoting neurogenesis.Overexpression of the encoded protein may be associated with astrocytoma progression.Neuroprotein 1 (NRN1) is involved in the PI3K-Akt-mTOR pathway,and studies have shown that NRN1 expression is frequently inhibited by methylation of promoter regions in human esophageal cancer cells.NRN1 was methylated in 50.4% of primary esophageal cancer samples,and NRN1 inhibited colony formation,cell proliferation,migration,and invasion,and induced apoptosis and G1/S arrest in esophageal cancer cells.NRN1 inhibits the growth of esophageal cancerin vitroandin vivoby inhibiting PI3K-Akt-mTOR signaling.Methylation of NRN1 is a novel synthetic lethal marker of PI3K-Akt-mTOR and ATR inhibitors in human esophageal cancer.
CREBBP or EP300
CREBBP genes play key roles in embryonic development,growth control,and homeostasis by combining chromatin remodeling with transcription factor recognition.
CREBBP proteins have intrinsic histone acetyltransferase activity and also act as scaffolds to stabilize the interaction of additional proteins with transcriptional complexes.It acetylates histones and nonhistones.The protein shares very high sequence similarity regions with protein p300 in its bromodomain,cysteinehistidine-rich region,and histone acetyltransferase domain.
The EP300 gene encodes an adenoviral E1A-associated cellular p300 transcriptional co-activator protein.It functions as a histone acetyltransferase and regulates transcription through chromatin remodeling and is important during cell proliferation and differentiation.It mediates cAMP gene regulation by specifically binding to phosphorylated CREB proteins.This gene has also been identified as a coactivator of HIF1A (hypoxia-inducible factor 1α) and therefore plays a role in the stimulation of hypoxia-inducible genes such as VEGF.Synthetic lethality using the mutation status of CREBBP/EP300 as a biomarker for the use of CARM1 small molecule inhibitors in DLBCL and other cancers.
WRN-MSI
DNA helicase WRN is a target for the synthesis of lethal cancer cells with microsatellite instability (MSI),a form of genetic hypermutation caused by impaired mismatch repair.Depletion of WRN induces extensive DNA double-strand breaks in MSI cells,leading to cell cycle arrest and/or apoptosis.WRN-encoded nuclear proteins are important in maintaining genomic stability and play roles in DNA repair,replication,transcription,and telomere maintenance.It contains an N-terminal 3‘to 5’ exonuclease domain,an ATP-dependent helicase domain,and an RQC (RecQ helicase conserved region)domain in its central region,as well as a C-terminal HRDC(helicase RNaseDC end) domain and nuclear localization signal.It preferentially binds DNA substrates containing alternative secondary structures,such as replication forks and Holliday junctions.It may play an important role in the dissociation of joint DNA molecules that can emerge as products of homologous recombination,replication fork stalling,or during DNA repair.It alleviates the arrest of DNA polymerase at the site of DNA damage.TAdinucleotide repeats are very unstable in MSI cells,and undergo large-scale expansion,which is different from insertion or deletion mutations of several nucleotides described previously.Extended TA repeats form non-BDNA secondary structures that stall replication forks,activate ATR checkpoint kinases,and require WRN helicase unwinding.In the absence of WRN,the extended TA-dinucleotide repeat is readily cleaved by the MUS81 nuclease,resulting in massive chromosome fragmentation.The synthetic lethal dependence of WRN in MSI and supports the development of therapeutic agents for WRN against MSI-associated cancers.
CYP2S1-BRAF
CYP2S1 gene is in the epidermis and may contribute to the oxidative metabolism of all-trans retinoic acid.For this activity,molecular oxygen is used to insert one oxygen atom into the substrate,and then the second oxygen atom is reduced to a water molecule,and the two electrons are passed by NADPH through cytochrome P450 reductase(NADPH-hemoprotein reductase).In addition,peroxidase and isomerase activities were shown for various oxygencontaining eicosanoids such as prostaglandin H2 (PGH2)and hydroperoxyeicosatetraenoate (HPETE).CYP2S1 is highly expressed in papillary thyroid carcinoma (PTC),especially in conventional PTC (CPTC) and high-cell PTC (TCPTC),and its expression is positively correlated with BRAF mutations.The BRAF-mediated MAPK/ERK cascade upregulates CYP2S1 expression through an AHR-dependent pathway,whereas CYP2S1 in turn enhances the transcriptional activity of AHR through its metabolites.This AHR/CYP2S1 feedback loop strongly amplifies the oncogenic role of BRAF in thyroid cancer cells,which leads to the fatal interaction between synthetic CYP2S1 and BRAF.
TET2
TET2 dioxygenase catalyzes the conversion of the modified genomic base 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC) and plays a key role in active DNA demethylation.5-hydroxymethylcytosine is favored in CpG motifs.It also mediates the subsequent conversion of 5hmC to 5-formylcytosine (5fC) and the conversion of 5fC to 5-carboxycytosine (5caC).The conversion of 5mC to 5hmC,5fC,and 5caC may be the first step in cytosine demethylation.Methylation at the C5 position of cytosine bases is an epigenetic modification of mammalian genomes and plays an important role in transcriptional regulation.In addition to its role in DNA demethylation,it is also involved in recruiting O-GlcNAc transferase OGT to CpG-rich transcriptional start sites of active genes,thereby promoting inactivating mutations in histone H2BGlcNAcylationTET2 through OGT is the initial genetic damage for hematopoietic stem and progenitor cell (HSPC) transformation,and the mechanism of selective killing of TET2 mutant blood cells is due to abnormally low levels of tyrosyl DNA phosphodiesterase 1 (TDP1),an enzyme important for the removal of TOP1 cleavage complex (TOP1 cc).Low TDP1 levels confer sensitivity to TOP1-targeting agents or PARP1 inhibitors,and are unable to remove the TOP1 cleavage complex,leading to DNA double-strand breaks and cell death.
IDH
Mutations in IDH 1 have been observed in a variety of cancer types,including sarcoma,hematologic malignancies,colon cancer,and brain cancer.Mutations in two isocitrate dehydrogenases involved in the conversion of α-ketopentanoic acid to D-2-hydroxypentanoic acid by the cytoplasm (IDH 1) and mitochondria (IDH 2) have been described as mutually exclusive in many of these cancer types.The most common mutations involve R132 (IDH 1) and R172 (IDH 2) involving the active site and resulting in new variant enzymatic activity.The impact of mutations in this gene varies by cancer type.In myelodysplastic syndromes and acute myelogenous leukemia (AML),IDH1 mutations are associated with worse outcome,shorter overall survival,and normal karyotype.However,in glioblastoma and astrocytoma,patients with IDH 1 mutations show better overall survival than patients with wild-type IDH 1.Unlike the association with cytogenetically normal AML,in glioblastoma,IDH 1 mutations are associated with specific cytogenetic abnormalities,1p and 19q deletions.Another approach to target IDH1 mutations is by inducing synthetic lethality of compounds targeted by poly (ADPribose) polymerase (PARP),glutamine metabolism,and the Bcl-2 protein family.
Summary and outlook
Tumor synthetic death may be one of the most important advances in modern cancer therapy.
In this paper,the mechanism of action of target genes in common tumors such as lung cancer,breast cancer,ovarian cancer,colorectal cancer,prostate cancer and other uncommon tumors and the principles leading to the occurrence of synthetic lethal effects are systematically discussed.The concept of synthetic lethality has great potential in anticancer drug discovery and may become an important means of inhibiting tumors at the genetic level in guiding cancer therapy in the future.During the initiation of oncogene program,inhibition of multiple signaling pathways and upstream and downstream molecules of signaling may fundamentally inhibit the development of tumors and drug resistance phenomena during treatment.We proposed the concept of synthetic lethality half a century ago,and cancer gene-targeted drug research in the 21st century was precisely based on NGS and CRISPR gene editing technologies.With the mining of CRISPR gene technology,more and more target genes are discovered in tumors,and humans inevitably face the application of synthetic lethality to inhibit tumors during anti-tumor.Whether some tumors can be accurately and efficiently cured in the future still has the way to go.In this paper,we enumerate the choice of drugs in common tumors under the theory of synthetic lethality,and more updated studies still need to be further validated and summarized.
Conflicts of interest
The authors indicated no potential conflicts of interest.
 Oncology and Translational Medicine2021年4期
Oncology and Translational Medicine2021年4期
- Oncology and Translational Medicine的其它文章
- Relationship between miR-7-5p expression and 125 I seed implantation efficacy in pancreatic cancer and functional analysis of target genes*
- The clinical efficacy of percutaneous ethanol-lipiodol injection (PEI) combined with high-intensity focused ultrasound (HIFU) for small hepatocellularcarcinoma in special or high-risk locations*
- Enhancing the treatment effects of tumor cell purified autogenous heat shock protein 70-peptide complexes on HER-3-overexpressing breast cancer *
- Efficacy and adverse reactions of apatinib as secondline or later-line treatment in advanced lung cancer
- Relationship between molecular changes in epidermal growth factor receptor (EGFR)and anaplastic lymphoma kinase (ALK)mutations in lung adenocarcinoma *
- The expression of vascular endothelial growth factor (VEGF)/ endostatin (ES) and VEGF receptor 2(VEGFR2)/ES is associated with NSCLC prognosis *