
DNA存储中的纠错方法综述
2021-09-10 05:55昝乡镇姚翔宇陈智华刘文斌
广州大学学报(自然科学版) 2021年2期
昝乡镇, 姚翔宇, 许 鹏, 陈智华, 谢 恋, 刘文斌*
(广州大学 a.计算科技研究院; b. 黄埔研究院, 广东 广州 510006)
随着分布式、云计算和物联网技术的发展,人类每天产生的数据总量呈现出指数增长的趋势.据国际数据公司(IDC)预测,2025年,全球数据信息总量将达到175 ZB[1].传统的磁、光、电等存储技术已无法满足未来呈指数增长趋势的数据存储需求[2-3].与此同时,作为生命信息存储介质的DNA分子在存储容量、稳定及能耗方面有着巨大的优势[4-5].据估计,DNA分子的存储密度可达到~107GB/mm3,比传统存储介质提高了6个数量级.正是基于上述原因,DNA分子有望成为解决海量大数据存储困境的一种极具潜力的存储介质[6-7].
20世纪60年代,Wiener[8]和Neiman[9]提出了用DNA分子存储数据的想法,标志着DNA存储研究的正式开始.随着合成和测序技术的成熟与推广,2012年,哈佛医学院的Church等[10]第一次在体外实现了0.65 MB数据的DNA存储.此后的每一年,《Nature》 《Science》等权威期刊都有相关研究成果的报道[10-14].欧美发达国家已经将DNA存储列入国家发展战略.2020年11月,微软公司与西部数据公司、Twist生物科技公司以及Illumina公司结成联盟,用于推进DNA数据存储领域的发展.美国半导体产业协会(SIA)发布的《半导体10年计划》也将DNA数据存储列为未来海量数据存储的重要选项.我国两会公布的《中华人民共和国国民经济和社会发展第十四个五年规划和2035年远景目标纲要》也明确指出,要加快布局DNA存储等前沿技术.
然而,DNA存储过程会不可避免地引入一些错误,这些错误对DNA数据的准确恢复提出了严峻的挑战.因此,DNA存储研究必须要解决的一个关键问题是如何在DNA数据恢复的过程中发现错误并纠正错误.本文主要介绍了DNA存储过程中错误的类型及其分布复杂性,DNA存储纠错技术的主要进展,并对其发展趋势进行了展望.
1 DNA存储信道复杂性
DNA存储过程主要包括4个步骤:将计算机数据编码成DNA序列,合成DNA序列,PCR扩增与存储,测序并恢复存储数据.DNA存储主要涉及3种生物技术:DNA序列合成技术、PCR扩增技术以及测序技术.DNA存储过程可以理解为一个信道模型,该信道模型主要由上述3种技术引起一些错误.这些错误可以分成两类:序列内错误和序列间错误.
序列内错误主要是指DNA分子序列存在碱基的插入、删除和替换等错误[15-17].DNA分子在合成时可能会发生碱基的替换、插入和删除等错误;PCR扩增阶段,可能发生替换错误;DNA分子在测序读取阶段,可能会发生碱基的替换、插入和删除等错误.研究表明,使用二代测序技术,每个碱基发生错误的概率为1%~2%[17], 而使用三代测序技术每个碱基发生错误的概率为10%~15%[18-19].此外,插入或删除错误会导致DNA序列的长度与标准序列长度发生偏差.实验表明,使用三代测序技术大约88%的reads长度不正确[20].
序列间错误主要是指DNA分子的缺失,以及DNA分子拷贝数分布不均匀[21].DNA编码序列在合成阶段,由于合成的不均匀性,DNA分子会被合成几百甚至几千个拷贝;DNA分子在PCR扩增阶段也会发生拷贝数不均匀的现象,甚至有些序列会直接丢失[22-23];在测序阶段,各个序列拷贝分布的不均匀性,将会导致测序数据中各序列对应的读长(reads)也存在不均匀性,甚至会丢失某些序列的读长.
可以看出,DNA存储过程中每个阶段都会发生DNA分子序列内的错误以及序列间的错误,这些错误相互叠加,增加了DNA存储解码过程的复杂性(图1).

图1 DNA存储信道的复杂性Fig.1 The complexity of DNA storage channel
有两个原因直接导致了DNA存储信道的复杂性:一个是技术原因,即技术水平存在缺陷;另一个是生物序列本身的一些约束没有遵循.生物序列约束主要包括GC含量、均聚物长度以及回文结构.DNA序列中GC分布不均匀、GC含量过低或者过高会导致DNA序列PCR扩增分布不均匀;长度大于4的均聚物的出现会导致DNA序列在合成或测序时发生碱基错误;DNA序列存在的回文子序列会导致该序列在PCR扩增时出现发夹等二级结构,进而影响PCR扩增的效率[24-25].
2 DNA存储纠错技术
目前,DNA存储纠错主要聚焦于两个方面,即设计遵循生物序列约束的编码策略以及鲁棒的纠错算法.
2.1 遵循生物序列约束的编码策略
编码DNA序列时遵循生物序列约束,有助于减少DNA存储过程中产生的错误,并提高解码效率[26].文献[27]建议编码DNA序列时遵循如下约束[27]:
(1)GC均匀分布且GC含量值介于40%~60%;
(2)均聚物的长度小于4;
(3)DNA序列不存在回文结构.
在设计遵循生物序列约束的策略方面,人们普遍采用随机化的策略.
Church等[10]使用二进制编码策略(即0用A或G表示,1用C或G表示)来控制GC含量,避免均聚物及回文序列的出现.该二进制模型比较简单,虽然能较好地满足生物序列约束,但是该方法编码效率不高.为了提高编码效率并遵循序列约束,Goldman等[28]及Bornholt等[29]开发了一种三进制的编码策略,即将数据转换为三进制数字0、1、2的表示形式,然后根据编码表将三进制数字转变成DNA碱基,见表1.Goldman等[28]设计的编码表,每个数字有多个对应的DNA碱基,当前数字对应的DNA碱基依赖于前一位数字确定的DNA碱基.然而,使用二进制和三进制模型编码DNA序列的研究比较少,目前的研究领域普遍采用四进制编码DNA序列,即两个二进制位对应一个碱基.

表1 三进制编码表
四进制编码策略容易出现GC含量过高及均聚物较多的问题.为了解决这些问题,目前主流的方法是先将二进制数据与一个符合要求的随机二进制序列做异或运算,形成一个二进制的结果序列,然后再对此二进制结果序列进行四进制编码[20, 30].
DNA喷泉码方法[26-27,31-32].通过随机选择二进制分组数据进行异或形成复合序列(液滴),然后将复合序列按四进制编码策略转换成DNA序列.如果DNA序列符合生物序列约束,则留下等待后续合成,否则直接舍弃并继续下一个复合序列(液滴)的生成.DNA喷泉码充分利用了喷泉码的无速率特性,省去了约束映射的冗余性.Wang等[33]采用混合的编码策略,即随机排列DNA序列里的碱基与可变长度映射策略相结合的方法,来满足GC含量及均聚物的约束.其中,可变长度映射策略的构建方法如下:①根据可变映射规则将不符合生物序列约束的二进制数据分组转换为codeword序列,例如,二进制序列‘1100-00-00-1101-01-111100-0’转换为‘01-1-1-02-2-001-1’;②将codeword序列每个数字与其前一个数字相加并对4求余,进而得到codeword序列对应的四进制序列,即‘01-1-1-02-2-001-1’转换为‘01-2-3-31-3-330-1’;③将四进制序列中的0映射为A碱基,1映射为T碱基,2映射为C碱基,3映射为G碱基,由此得到符合生物序列约束的DNA序列,即‘01-2-3-31-3-330-1’编码成DNA序列‘AT-C-G-GT-G-GGA-T’.需要说明的是,上述步骤中可变映射规则的构建是通过构建一个包含3个状态、4个数字(代表DNA的4个碱基),且连续出现的0不能超过2的状态转换图FSTD(4,0,2),进而进行Huffman编码得到的,见图2.
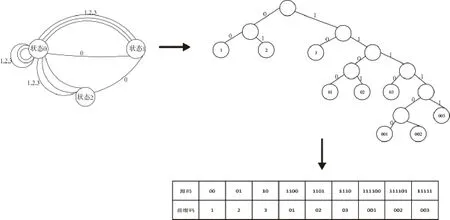
图2 可变长度的映射规则Fig.2 Variable-length constrained mapping rule
Zhang Yi团队[13]、Yazdi等[34]及Xue 等[35]采用一种将用户二进制数据与满足0、1均衡的给定二进制序列(调制码序列)进行调制的思想,来满足GC均衡及均聚物的约束.具体方法为将待编码二进制序列与调制码序列逐列对齐,然后将每列按照调制规则转换成一个DNA碱基,进而得到该二进制序列调制后的DNA序列(表2).
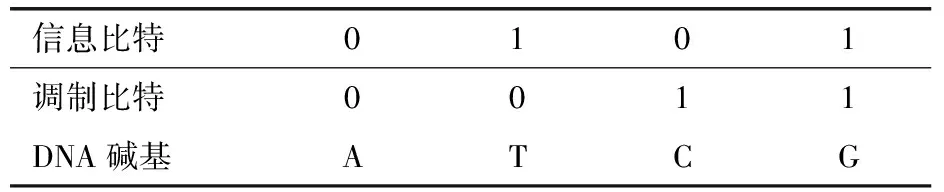
表2 调制规则
需要指出,上述所有方法除Church等[10]的方法外,均不能完全解决编码序列存在二级结构的问题.
2.2 DNA序列数据纠错技术
2.2.1 序列丢失纠错
目前,研究人员主要通过在编码阶段增加冗余序列来解决DNA存储中序列的丢失问题.3种代表性的异或方法如图3所示.
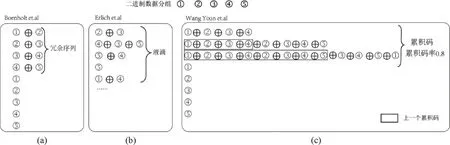
图3 3种代表性的异或方法Fig.3 Three typical XOR methods
根据添加冗余序列方式的不同,DNA序列丢失的解决方法可以分为两类.第一类主要是通过“序列异或”的思想产生冗余序列或复合序列.Bornholt等[29]采用连续两个二进制序列进行异或产生第三个冗余序列的方法来解决丢失(图3a).采用这种方法,任意一个序列丢失,可以通过该序列相关联的其他两个序列异或得出.然而,该种方法序列冗余度比较大.
为了降低冗余,2017年,Erlich等[27]在喷泉码思想的基础上提出了DNA喷泉码算法(图3b).其方法为①随机选择多个二进制数据分组进行异或操作生成复合序列(也叫液滴);②合成并存储复合序列(液滴);③PCR扩增并测序每个复合序列;④将已经分解出的二进制分组依次异或包含该二进制分组的液滴,直至分解出所有二进制分组数据.尽管该方法可以以很高的DNA逻辑存储密度(1.57 bit/nt)复现数据,但是该方法编码和解码的复杂度与数据大小并不是线性相关,相比编码解码更复杂.Ping等[36]认为尽管Erlich等宣称DNA喷泉码丢失4%的液滴(指DNA编码合成的序列)不会影响源数据的恢复,但是丢失更多的数据将有可能导致整个源数据无法恢复.因此,存储永久性的数据就必须有足够量的冗余.
考虑到DNA喷泉码中潜在的解码失败和冗余地址(降低净信息密度)可能会阻碍其在DNA扩展性存储中的实际应用,Wang等[33]提出了累积码的概念,即在源文件二进制分组数据的基础上,添加一定量的冗余校验序列.冗余校验序列生成的方法为(图3c):根据累积码率,选择一定量的二进制分组数据(注意:图例中选择二进制分组的数量为5*0.8=4),将其与前一个冗余校验序列一起进行异或运算.使用该方法,可以通过冗余校验序列和丢失序列相关的其他二进制序列进行异或运算,即可恢复丢失的序列.
第二类通过使用纠错码(Error Correcting Codes, ECCs)来实现缺失序列的恢复.该类方法主要是通过在一个数据块内添加一些冗余序列(又叫外码)来实现,参见图4.基本思想是通过将缺失序列的位信息转变成替换错误,然后通过纠错码来实现纠正[20, 37-38].具体方法是①首先将数据块内的二进制数据分组按列排放,形成一个矩阵;②依次向矩阵每一行的左侧(或右侧)使用纠错码添加r位冗余位信息.解码时先将数据块内的二进制数据根据各自的Index值依次排列好,缺失序列用等长的随机二进制数据替换,然后依次对矩阵的每行使用纠错码纠正缺失序列对应的位信息,直至缺失序列完全恢复为止.考虑到目前DNA合成技术一次最多可以同时合成DNA链的数量为224=16 777 216,Meiser等[37]最先针对图4框架中一个数据块内二进制分组数据的个数k、二进制分组数据的大小(包括分组数据数据块内的序号ID)、外码的个数以及每个符号所占位数等参数的组合进行了研究,并给出了每种参数组合下每条编码序列包含的碱基个数.目前,该类方法使用的纠错码主要为Reed-Solomon码(RS码).

图4 外码和内码框架Fig.4 The framework of outer code and inner code
2.2.2 序列内碱基错误纠错
(1)基于多序列比对思想上的纠错
前面已经指出,每个DNA序列在DNA存储信道中都会有多个拷贝.研究人员利用DNA序列的多拷贝性来纠正DNA序列内的碱基错误,即替换、插入和删除等错误.该算法实现纠错不要求参与比对的读长具有相同的长度.该算法的基本思想是,把属于同一序列的多个拷贝聚类(或分组)到一起,然后使用多序列比对软件(比如MUSCLE,MAFFT等[39-40])比对同一序列的多个拷贝,然后按照多数投票原则确定出一个一致性的序列,见图5.该类方法可以纠正序列内碱基的插入、缺失和替换等错误.
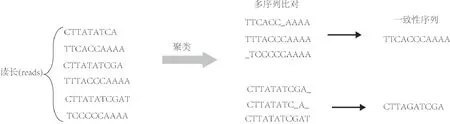
图5 聚类和多数投票示例Fig.5 Schematic diagram of clustering and majority voting
Goldman等[28]采用4倍重叠冗余来解决序列的碱基错误问题,即每个序列产生3个重叠的冗余序列,各个冗余序列和前一序列有3/4的重叠.然而该方法的DNA逻辑存储密度只有0.33 bit/nt,难以实际推广.为了降低合成成本,李彦敏等[41]没有使用重叠片段的编码思想,而是通过在序列内增加序列索引重复的方式提高后续聚类的精度,进而实现纠错.但是,该方法只能适应错误率较低的情形.
Yadiz等[34]纠正序列内碱基错误的方法分成两个阶段,如图6所示.第一阶段,根据每个读长(reads)的索引值(序列号)对reads进行分组,然后使用多序列比对软件对每个分组包含的读长进行比对,然后使用多数投票算法得到一致性序列(码字);第二阶段,迭代使用BWA[42]比对算法以及均聚物查找算法,修正第一阶段产生的一致性序列(码字).需要说明的是,第一阶段生成一致性序列的方式是通过逐个确定均聚物的方式而产生的.该方法完成纠错需要较高的测序深度(200X).
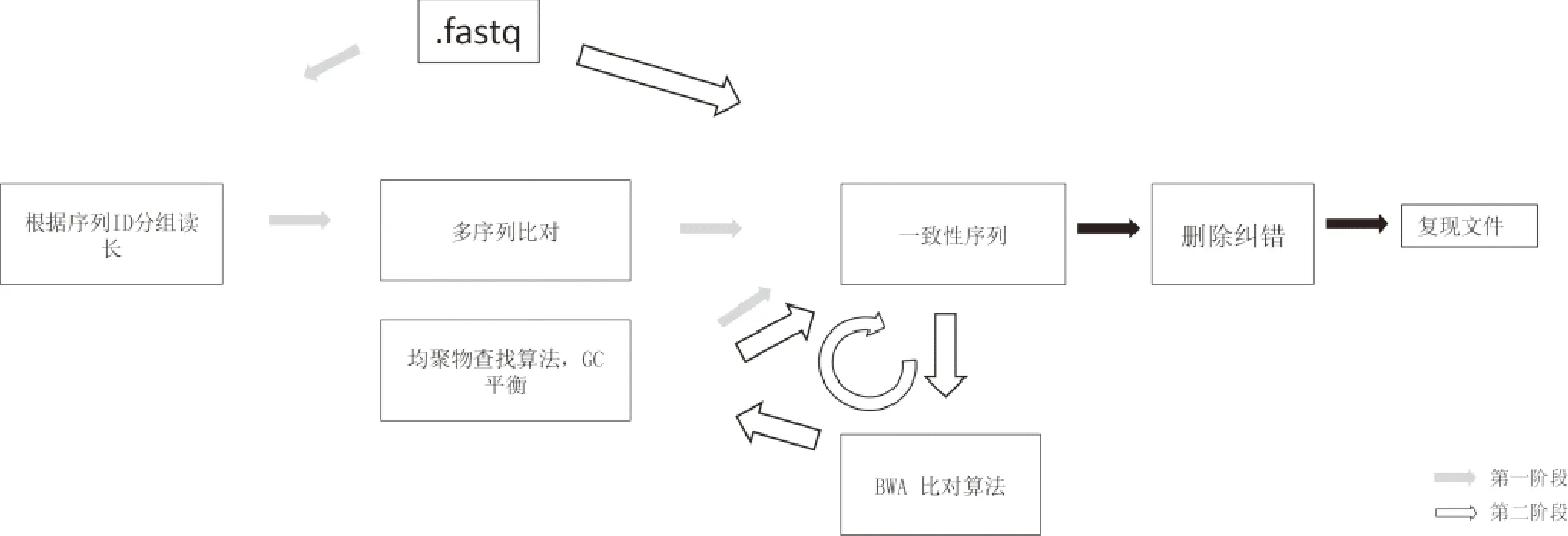
图6 序列比对及均聚物校正示意图Fig.6 Sequence alignment and homopolymer correction
Organick等[20]、Antkowiak等[30]以及Jeong等[26]通过改进聚类算法,提高了产生一致性序列的准确度. Organick等[20]、Antkowiak等[30]使用局部敏感哈希办法聚类测序得到读长(reads),该种聚类方法相比以汉明距离为基础的Jaccard相似性聚类方法,具有更快的时间性能.该方法的基本思想是,首先获取各读长在给定随机数矩阵上的签名,然后通过比较签名的相似性完成读长的聚类.Jeong等[26]首次将序列各碱基的Q-score值纳入聚类算法考虑的因素.
(2)基于传统通信纠错码上的纠错
传统通信纠错码可以纠正二进制数据分组上的错误,尤其擅长纠正替换错误,且具有完备的数学理论基础.由于DNA存储信道和传统通信信道相比,都是将计算机二进制数据经过一系列变换最终再转变成二进制数据,这就使得用传统通信纠错码去纠正二进制分组数据里的错误成为可能.
目前,纠正DNA序列内碱基替换错误的方法,主要采用通信领域里性能较好的纠错码来实现检错和纠错的目的.这些纠错码包括Reed-Solomon码(RS码)[37, 43-44]、BCH码[45-46]、Raptor Code码[47]、汉明码[12, 48]、LDPC码[44, 49-50],等等,其中,主流的是使用RS码(图4中的内码是指RS纠错码).使用这些纠错码的方法是①针对二进制分组数据,使用上述纠错码对应的生成矩阵产生冗余位信息;②将冗余位信息融入到二进制分组形成待编码二进制序列;③将待编码二进制序列按照四进制编码策略转换成DNA序列;④在将测序得到的DNA序列转换为二进制序列后,使用纠错码的一致性校验矩阵进行矩阵运算,从而检错和纠错.然而,上述纠错码纠错的能力越强,即纠错的数量越多,冗余度就越高.例如,BCH(255,47)码可以纠正替换错误率达16%的错误,但是冗余度达到82%.
Xue等[35]在Levenshtein码的基础上,开发了一个既能实现GC平衡,又能解决碱基插入/缺失/替换错误的编码方法.该方法构建levenshtein码的方法为(图7)①将每一个长度为(2n-3「log2n⎤-2)的二进制分组数据拆分成长度为n和长度为(n-3「log2⎤n-2)的两个子串;②从最后一位开始,将长度为n的二进制分组数据子串逐位取反直至该子串含1的数量为n/2,标记为奇串;③将奇串的统计信息、校验信息以及长度为(n-3「log2n⎤-2)的二进制分组数据子串合并,构成偶串;④将奇串和偶串调制成DNA序列.解码时,当有一位插入/缺失/替换错误发生时,奇串和偶串的对应信息会出现冲突,此时根据奇串的长度与标准长度的差值以及当前解码出的二进制字符串的S值和H值,可以准确地推算出发生碱基错误的位置并给予纠正.然而,该方法一个码字只能解决一位插入/删除/替换错误,且码字较长.
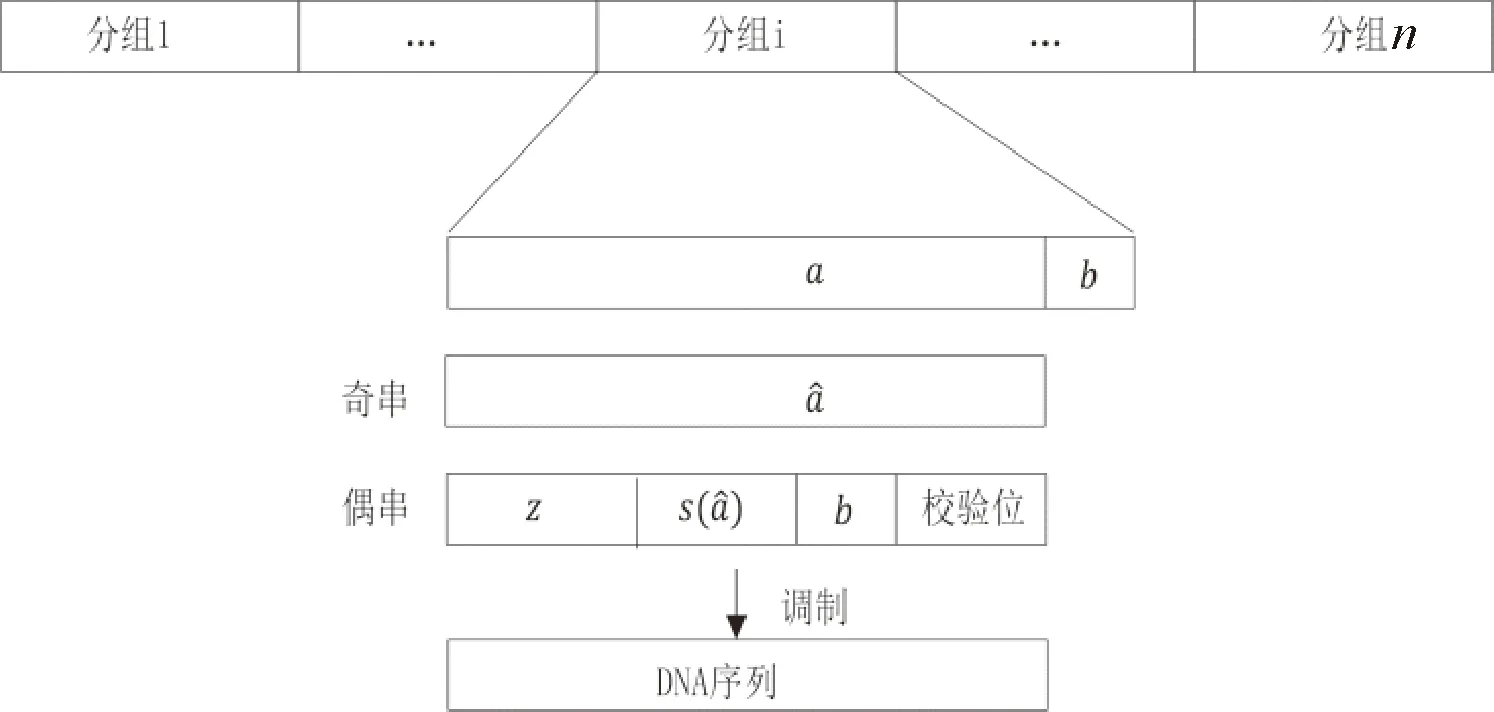
图7 Levenshtein码的构建Fig.7 Construction of Levenshtein code
(3)基于算法角度实现纠错
纠正DNA序列内错误的难点是纠正序列里的插入和删除等错误,因为很难分辨出DNA序列内具体哪些碱基发生了这些错误.因此,纠正错误的关键在于准确地识别出发生插入/删除错误的位置.目前,研究人员大都通过设计一套有一定规律的编码策略来编码DNA序列.该编码策略对插入/删除非常敏感且不需要额外的碱基进行存储.
Blawat等[46]设计了一套编码表,即每个字节对应两个DNA编码(图8a),然后将所有字节的DNA编码分成两类,编码DNA序列时,交替使用各字节的第一类编码和第二类编码.当插入/缺失错误发生时,第一类DNA编码和第二类DNA编码交替出现的规律将会打破,由此可以定位发生插入/删除的编码位置(图8b).然而该算法只能检测插入/缺失错误,不能纠错.

图8 Blawat设计的编码表及可检测插入/缺失错误的示例Fig.8 The code table by Blawat and an illustration of insertion/deletion detecting
Press等[51]设计了一个名为HEDGES的算法,同时结合RS码,用于纠正DNA序列内的插入/删除/替换错误(图9).其方法:①将源文件二进制数据划分成等长度的二进制信息,然后对每个二进制信息添加索引(即序列ID),形成一系列待编码二进制序列;②将待编码二进制序列的每个二进制位数值、位置下标索引、序列index以及该位前面连续几个位信息进行hash操作最终变成一个DNA碱基,由此得到一个待编码二进制序列对应的DNA序列.当插入/删除/替换错误发生时,上述编码规律将会打破,通过查找树结构可以判断该二进制位是否发生错误及发生什么错误,并最终将出错二进制位信息予以纠正.实验结果表明,该方法能够处理约1.2%的增删错误.然而该方法的编码率不高,纠正3%的错误,编码率将降低到0.6以下.
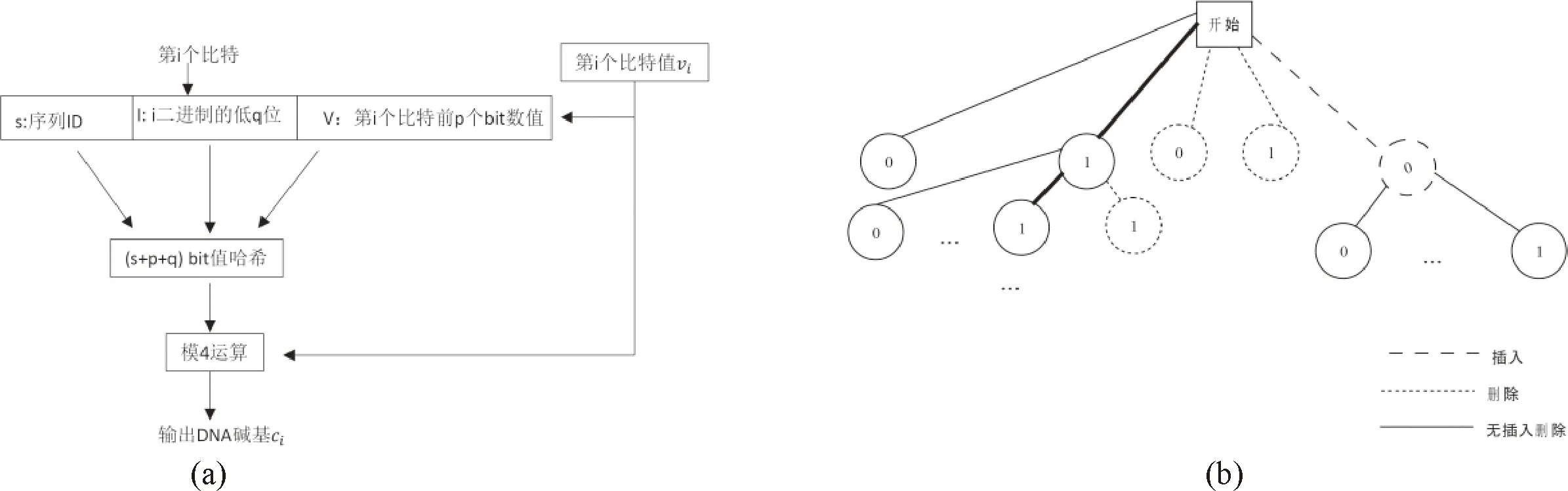
图9 HEDGES编码与解码Fig.9 Schematic diagram of HEDGES encoding and decoding
天津大学的Song 等[52]在De Bruijn图的基础上,构建了同一序列多个拷贝的De Bruijn图,然后通过基于贪心的搜索策略,搜索原序列对应的最可能路径.该算法与第一类算法相比,不需要聚类,因此具有更快的时间性能(图10).该方法可以解决序列内的插入、删除、替换等错误.但是,该方法De Bruijn图的构建过度依赖于序列索引(即序列ID).如果属于同一序列的每个拷贝index都出现错误,该方法将不能构建De Bruijn图.

图10 de Bruijn 图算法框架Fig.10 The framework of de Bruijn graph
Sharma等[53]通过定义碱基异或规则(表3)实现了纠正碱基替换错误的目的.其方法是①将每一个DNA编码序列从左到右4个碱基为一组切分该编码序列;②依次将每个切分第一字符拼接起来形成第一个编码序列,同时将每个切分后续3个字符拼接起来形成第二个编码序列;③依次将每个切分第一个字符与该切分后续3个字符做异或操作,将所有切分异或操作结果依次拼接起来,形成第三个编码序列;④解码时,根据得到的属于同一序列的3个片段,两两异或推导出3个原编码序列,根据一致性最终确定原编码序列.然而,该方法的冗余度比较高,冗余度为75%.

表3 碱基异或表
3 DNA纠错技术展望
DNA数据存储是一种新兴的非易失性存储技术,具有前所未有的密度、耐用性和复制效率.尽管DNA存储与传统存储技术相比,具有较明显的优势,但是现阶段DNA存储距离真正的大规模应用还有很长的距离.未来DNA存储要做到真正大规模的应用,使用成本较低、通量较大,但同时错误率较高的DNA合成技术及DNA测序技术再所难免.因此,未来DNA存储纠错技术可能呈现出以下两个变化.
(1)编码DNA序列时严格遵循生物序列约束.在满足GC平衡及无均聚物的同时,尤其要避免DNA二级结构的形成.因此,未来的纠错算法可能包括DNA二级结构子序列的查找算法以及DNA二级结构的替换算法,从源头上严格保证DNA编码序列满足生物序列约束,尽可能减少后续DNA存储过程中的错误.
(2)未来的DNA存储纠错算法应能适应错误率更高的环境,同时具有良好的时间性能.目前的DNA存储纠错算法能够适应的碱基错误率最高为15%左右(使用三代测序).随着人们对DNA存储成本的要求越来越低及对存储通量的要求越来越高,未来DNA存储纠错算法可能要适应错误率高达20%以上的环境.针对高噪声环境,设计鲁棒的纠错算法,同时满足处理海量DNA数据时间性能上的要求,是未来DNA存储纠错算法的方向.
猜你喜欢
中等数学(2021年8期)2021-11-22
教学考试(高考生物)(2020年6期)2020-11-23
食品与生物技术学报(2020年8期)2020-01-06
数学大王·低年级(2019年10期)2019-11-25
中等数学(2019年4期)2019-08-30
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
小学生学习指导(低年级)(2019年3期)2019-04-22
小学生学习指导(低年级)(2018年9期)2018-09-26
小学生导刊(低年级)(2017年1期)2017-06-12
