
弥漫内生性脑桥胶质瘤靶向治疗新策略的体外筛选与验证
2021-09-09 08:09:46韩雨洁唐玉杰
上海交通大学学报(医学版) 2021年8期
李 瑞,韩雨洁,张 蕾,唐玉杰
上海交通大学医学院病理生理学系,细胞分化与凋亡教育部重点实验室,上海 200025
肿瘤是目前14岁以下儿童死亡的第二大原因,仅次于意外[1]。中枢神经系统肿瘤已经超过白血病成为发生率和致死率均列第一的儿童肿瘤类型[2]。弥漫内生性脑桥胶质瘤(diffuseintrinsic pontine glioma,DIPG)发生在儿童脑干的脑桥区域,中位生存期约为9个月,2年生存率低于10%,5年生存率不足1%,是目前已知致死性较高的肿瘤类型[3-4]。由于DIPG发生的脑桥区域存在控制心率和呼吸的重要神经,而肿瘤本身又具有很强的浸润性,因此临床上该类肿瘤无法通过手术来治疗[5]。目前仅放射治疗(放疗)能够发挥一定的作用,但治疗后肿瘤会很快复发并形成放疗耐受,因此亟需发现针对DIPG的有效治疗手段[6]。
研究[7]发现约80%的DIPG患者携带H3组蛋白编码基因H3F3A(H3-3A gene)或HIST1H3B(H3 clustered histone 2)的体细胞突变,使得第27位赖氨酸突变成甲硫氨酸,即H3K27M突变。虽然DIPG中H3K27M的表观遗传肿瘤驱动功能已经被业内公认,但是该突变很难被小分子直接靶向,因此近年来包括笔者所在团队在内的国内外多个团队都聚焦在鉴定能够间接拮抗H3K27M致癌功能的表观遗传靶向策略,并报道了溴结构域和外端家族(bromodomain and extra terminal protein,BET)抑制剂[8]、细胞周期蛋白依赖性激酶7(cyclin-dependent kinase 7,CDK7)抑制剂[8]以及组蛋白去乙酰化酶(histone deacetylase,HDAC)抑制剂[9]等表观遗传靶向小分子能够在临床前肿瘤模型中有效治疗DIPG,开拓了DIPG的表观遗传靶向治疗新思路。其中HDAC抑制剂panobinostat已获美国食品药品监督管理局(FDA)批准用于多发性骨髓瘤的治疗[10],多个BET抑制剂和CDK7抑制剂已进入肿瘤治疗的早期临床试验[11]。然而,笔者所在团队发现体外长期亚致死剂量药物作用可以诱导DIPG细胞产生针对panobinostat的获得耐药性;BET抑制剂处理DIPG细胞主要产生增殖抑制作用,并不诱导显著的细胞毒作用[8],这些都将限制其潜在的临床应用。
存活蛋白(survivin)由BIRC5(baculoviral IAP repeat containing 5)基因编码,是凋亡抑制蛋白(inhibitor of apoptosis,IAP)家 族 成 员[12]。肿 瘤 中survivin的高表达通常与化疗耐药、转移风险增加、肿瘤复发的高风险相关,使其在多种肿瘤中被发现可作为能反映较差预后的生物标志物[13]。同时survivin靶向抑制剂YM155也进入了临床试验[14-15]。
本研究结合转录组分析和药物库筛选寻找DIPG可能的靶向治疗策略,并进行体外验证,旨在为后续体内验证和机制挖掘奠定基础。
1 材料与方法
1.1 材料与试剂
1.1.1 细胞 DIPG原代细胞SU_DIPG13和SU_DIPG17由美国斯坦福大学Dr.Monje赠予,均来自H3.3组蛋白K27M突变的DIPG患者,经原代培养后可在体外稳定连续传代培养。
1.1.2 细胞培养基 DIPG培养基成分包括:Neurobasal-A培养基(10888022,美国Gibco)、DMEM/F12培养基、HEPES缓冲溶液(15630-080,美国Invitrogen)、丙酮酸钠溶液(11360-070,美国Invitrogen)、非必需氨基酸(11140-050,美国Invitrogen)、GlutaMAX-I(35050-061,美国Invitrogen)、B-27(12587-010,美国Invitrogen)、人表皮生长因子(AF-100-15-100,美国PeproTech)、人成纤维细胞生长因子(100-18B-100,美国PeproTech)、人血小板源生长因子AA(100-13A-50,美国PeproTech)、人血小板源生长因子BB(100-14B-50,美国PeproTech)、肝素(07980,加拿大StemCell Technologies)、抗菌抗真菌剂(15240-096,美国Invitrogen)。
小鼠神经干细胞(mouse neural stem cell,mNSC)培养基成分包括:鼠源神经干细胞培养基(NeuroCult NS-A proliferation kit mouse)(Cat05702,加 拿 大StemCell Technologies)、鼠表皮细胞生长因子(315-09-100,美国PeproTech)、鼠碱性成纤维细胞生长因子(450-33-50,美国PeproTech)、肝素、抗菌抗真菌剂。
1.1.3 其他试剂 筛选所用药物库部分由美国儿童癌症治疗发展研究所Dr.Charles Keller赠予,部分从美国Cayman Chemical和Selleck公司购买。Matrigel、多聚右旋赖氨酸(poly-D-lysine,PDL)、PBS(GP200603,武汉赛维尔生物科技有限公司),HBSS、蛋白裂解液、TRIzol、TrypLE、DNA酶、CellTiter-Blue(G8080,美国Promega),CellTiter-Glo(G9243,美国Promega),BCA蛋白定量试剂盒、反转录试剂盒、SYBR Green、EdU试剂盒、凋亡试剂盒、SDS-PAGE凝胶(上海熠晨生物科技有限公司),survivin抗体(71G4B7,美国Cell Signal Technology)。死亡结构关键结构域BH3的模拟抑制剂ABT-737、酪蛋白激酶2(casein kinase 2,CK2)抑制剂CX4945(美国Selleck)。
1.1.4 小鼠 出生后48 h的BALB/c小鼠2只,饲养于上海交通大学医学院实验动物科学部SPF级动物房。动物使用许可证号SYXK(沪)2018-0027,生产许可证号SCXK(沪)2018-0007。所有实验动物相关操作均获得上海交通大学医学院实验动物使用和管理委员会批准。
1.2 实验方法
1.2.1 细胞培养 DIPG原代细胞37℃培养箱常规培养。mNSC从出生后48 h的BALB/c小鼠小脑获取。2只小鼠断头处理,取出小脑,去脑膜,分离皮层,培养皿中切碎后用TrypLE在37℃振荡消化11 min,离心后HBSS重悬,40μm滤网过滤,离心后mNSC培养基重悬培养,0.1 mg/mL的PDL包被孔板37℃放置2 h,蒸馏水洗净后用于mNSC培养。
1.2.2 DIPG转录组数据分析 8例DIPG肿瘤组织以及6例对照远端正常脑组织的mRNA测序(mRNA sequencing,RNA-seq)数据来自前期发表的文章[9]。R包DESeq2用于分析差异表达基因(differentially expressed genes,DEGs),以调整P值(adjustedPvalue,padj)≤0.05、|log2差异倍数(fold change,FC)|≥1为标准筛选。R包ggplot2绘制散点图、火山图,R包pheatmap绘制热图。基因富集分析(Gene Set Enrichment Analysis,GSEA)进行基因本体(Gene Ontology,GO)和通路(Pathway)分析。GO分析包括生物过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF);Pathway分析包括京都基因与基因组百科全书(Kyoto Encyclopedia of Genesand Genomes,KEGG)、BioCarta和REACTOME。
1.2.3 靶向小分子单药和组合筛选 根据转录组分析并结合DIPG和其他脑胶质瘤的已有靶向治疗研究成果,选择若干靶向小分子进行DIPG原代细胞的杀伤筛选和验证。选择一些已经在DIPG中报道过的靶向小分子作为阳性对照,包括靶向小分子THZ1、panobinostat、AUY922、PR-171、flavopiridol等[8-9,16]。
设置0.01、0.1、1、10μmol/L 4个筛选浓度,以不做处理的空白组(MOCK)和二甲基亚砜(dimethyl sulfoxide,DMSO)处理组为阴性对照,每个浓度设置3个复孔。384孔板每孔铺板1 500个SU_DIPG13细胞,加药处理72 h,CellTiter-Blue检测细胞活力。
同时用以上靶向小分子在0.01、0.1、1、10μmol/L浓度下与BET抑制剂JQ1或HDAC抑制剂panobinostat进行组合筛选。JQ1和panobinostat的筛选浓度为40%抑制浓度(inhibitory concentration,IC),即IC40。同样以不做处理的MOCK组和DMSO处理组为阴性对照。
1.2.4 药物反应曲线测定 对于要体外单独验证的药物,分别选择5个药物浓度(0.000 1、0.001、0.01、0.1、1μmol/L)绘制药物反应曲线。96孔板每孔铺板5 000个SU_DIPG13细胞,每个浓度条件设置3个复孔,加药72 h后CellTiter-Glo检测细胞活力,用GraphPad Prism 8软件绘制药物反应曲线。
1.2.5 Western blotting 药物处理SU_DIPG13细胞48 h后,收集细胞悬液至EP管中,离心,弃上清液,沉淀用含有苯甲基磺酰氟(phenylmethylsulfonyl fluoride,PMSF)的蛋白裂解液吹打,并在冰上裂解30 min,离心。收集上清液至新的离心管中,BCA试剂盒检测蛋白浓度并定量。配制SDS-PAGE凝胶,每个样品上样10μg,100 V电压条件下进行电泳,直至溴酚蓝迁移至胶底部。聚偏氟乙烯(polyvinylidene fluoride,PVDF)膜用甲醇活化后转膜,90 min后取出,用5%牛血清蛋白(bovine serum albumin,BSA)摇床封闭1 h后,加一抗(1∶1 000),于4℃摇床中过夜。微管蛋白为内参。TBST洗膜后,二抗(1∶5 000)室温孵育2 h,TBST洗膜后显影。
1.2.6 实时定量PCR 药物处理24 h后,收集SU_DIPG13细胞悬液至EP管中,离心,弃上清液,500μL TRIzol吹打至沉淀完全溶解;每管加入100μL氯仿,快速混匀,静置2 min;12 000×g、4℃离心15 min,吸取上清液至新的EP管中;每管加入2μL糖原、250μL预冷异丙醇,混匀静置2 min;12 000×g、4℃离心10 min;弃上清液,加入1 mL预冷75%乙醇,7 500×g、4℃离心2 min,重复此步骤;加20~30μL DEPC水溶解沉淀,定量。反转录试剂盒将RNA反转录为cDNA,SYBR Green实时荧光定量PCR(real time fluorescent quantitative PCR,qRT-PCR)试剂检测RNA表达水平。BIRC5和GAPDH引物序列如下:BIRC5上游引物5'-AGGACCACCGCATCT CTACAT-3',下游引物5'-AAGTCTGGCTCGTTCTCAGT G-3';GAPDH上游引物5'-TGACTTCAACAGCGACAC CCA-3',下游引物5'-CACCCTGTTGCTGTAGCCAAA3'。
1.2.7 药物对细胞增殖和凋亡的影响 设置MOCK、DMSO处理组作为对照组,设置药物处理组为实验组,分别检测SU_DIPG13细胞的增殖和凋亡水平。在药物处理细胞18 h后,实验组和对照组均加入EdU,使其终浓度为10μmol/L,37℃孵育6~8 h后收集细胞并消化为单细胞,4%多聚甲醛固定并破膜处理,APC荧光染色后,避光室温孵育30 min,流式细胞仪(CytoFLEX S,美国Beckman Coulter)检测细胞增殖活性。药物处理细胞48 h后,收集实验组和对照组细胞,并消化细胞为单细胞后,Annexin V和碘化丙啶(propidium iodide,PI)同时对细胞染色,避光室温孵育5 min,流式细胞仪检测细胞凋亡水平。
1.2.8 靶向小分子的组合验证 组合的2个靶向小分子各选择4个逐步升高的浓度分别进行单药和组合药物抑制率检测,并利用CalcuSyn 2.0软件计算组合指数(combination index,CI)。CI<1、CI=1、CI>1分别表示2个靶向小分子在对应浓度下存在协同抑制、叠加抑制、拮抗肿瘤细胞生长的效果[17]。
2 结果
2.1 DIPG肿瘤组织的转录组分析
为了鉴定新的DIPG靶向治疗策略,利用课题组前期已经发表的8例DIPG肿瘤组织和6例对照远端正常脑组织的RNA-seq数据进行DEGs分析[9],筛选出2 319个DEGs,包括1 020个在肿瘤中显著上调和1 299个在肿瘤中显著下调的基因(图1A、B)。对上述基因分别进行GO分析和Pathway分析,用于鉴定富集了表达失调转录本的致癌或抑癌相关生物学功能或信号通路(图1C、D)。DIPG中显著上调的基因主要存在于细胞外基质和细胞表面,参与微管、细胞外基质等形成和功能;显著下调的基因主要存在于突触上,功能上主要参与突触信号以及细胞之间的信号转导和神经分化。
2.2 DIPG原代细胞的靶向小分子筛选
以DIPG肿瘤转录组分析结果为基础,共挑选出10类66个靶向小分子(图2A)。这10类分子包括BET抑制剂、HDAC抑制剂、组蛋白甲基转移酶抑制剂(histone methyltransferaseinhibitor,HMTi)、组蛋白去甲基化酶抑制剂(histone demethylase inhibitor,HDMi)、细胞周期类抑制剂(cell cycle inhibitor,CCi)、代谢类药物(metabolic drug,MD)、激酶抑制剂(kinase inhibitor,KI)、mTOR抑制剂(mTOR inhibitor,mTORi)和通路抑制剂(signaling pathway inhibitor,SPi)等。
选择DIPG原代细胞SU_DIPG13对上述66个靶向小分子进行体外细胞活性抑制筛选。针对每个靶向小分子,分别测试了药物在4个浓度下处理SU_DIPG13细胞72 h之后对细胞活性的影响(图2B)。结果显示,在0.01、0.1、1、10μmol/L浓度条件下抑制率超过50%的靶向小分子分别有2、3、11和22个。最终以1μmol/L浓度下对细胞生长抑制率大于50%为筛选标准,获得符合该标准的11个靶向小分子;其中包含5个阳性对照小分子。根据抑制率大小,从高到低分别是YM155、AUY922、THZ1、CDK1/2 inhibitorⅢ、dinaciclib、PR-171、panobinostat、flavopiridol、LY2874455、INK128和JIB-04(图2C)。11个小分子中除了THZ1和JIB-04,均已进入临床试验(表1);而CDK7抑制剂THZ1有同类小分子药物进入Ⅰ期临床试验(NCT04247126)。
2.3 BIRC5靶向小分子药物YM 155对DIPG的抑制作用
在11个小分子的靶基因中,BIRC5、CDK2、CDK4、PSMB8、PSMB9在DIPG中特异性高表达(log2FC≥1,padj≤0.05)。其中只有靶基因为BIRC5的小分子YM155尚未在DIPG中报道过,并且与正常脑组织转录组比较BIRC5在DIPG中上调的倍数最高(log2FC=4.64,P=0.018;图2D)。此外,YM155杀伤SU_DIPG13细胞的IC50最小,且在1μmol/L浓度下的抑制效果也最强(表1)。因此,接下来对BIRC5基因进行后续验证。
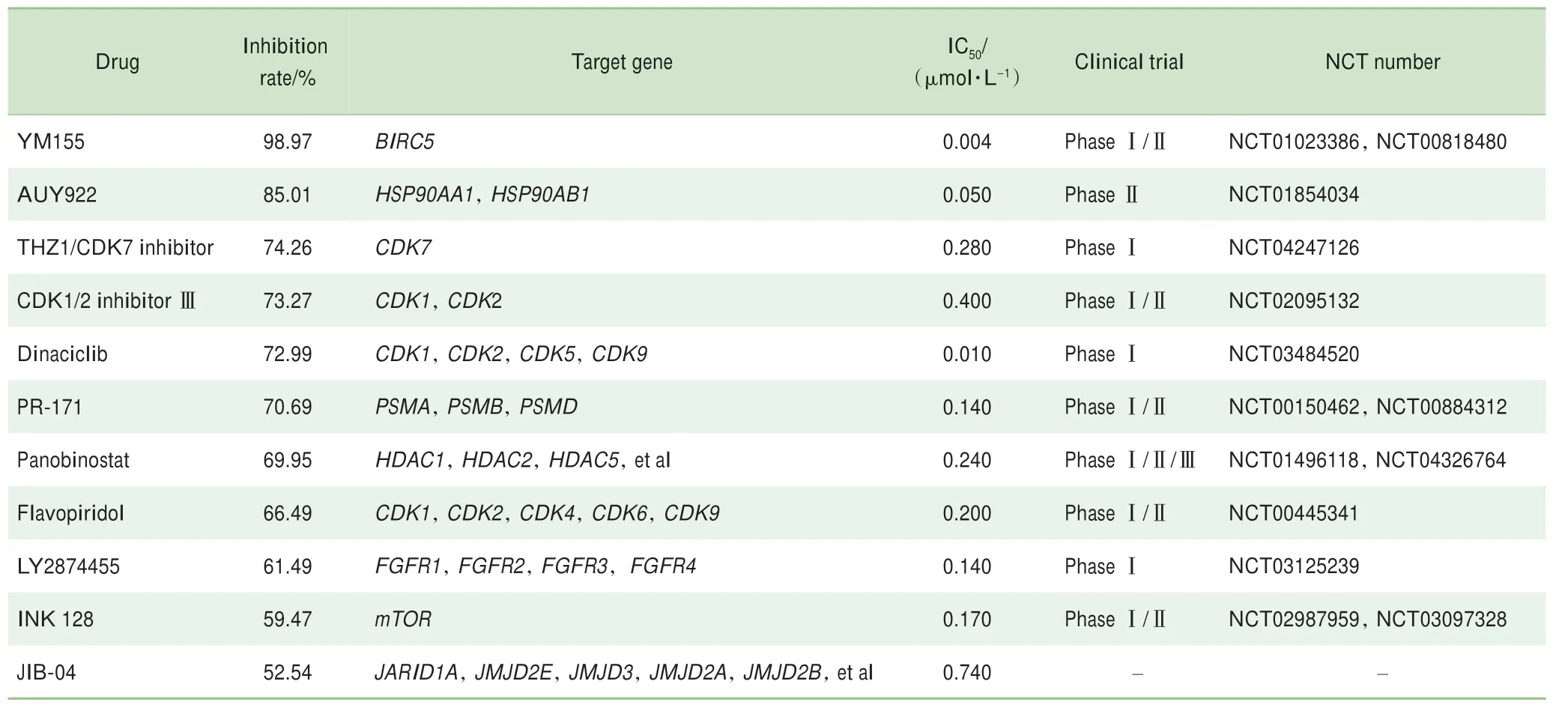
表1 在1μmol·L-1条件下对SU_DIPG13细胞抑制率大于50%的药物Tab 1 Drugs with growth inhibition rate over 50%at 1μmol·L-1 in SU_DIPG13 cells
首先通过qRT-PCR和Western blotting检测YM155在体外对BIRC5表达的影响。YM155处理SU_DIPG13细胞后BIRC5的表达在mRNA和蛋白水平均受到明显抑制(图3A、B)。平行比较YM155在2个DIPG原代细胞SU_DIPG13、SU_DIPG17和对照细胞mNSC的浓度-抑制率曲线后,2个DIPG原代细胞的IC50(0.004、0.007μmol/L)显著低于mNSC(0.03μmol/L),YM155在体外对DIPG细胞有更显著的抑制作用(图3C)。进一步通过EdU和Annexin V/PI标记染色的方法分别测试YM155处理DIPG原代细胞后对其增殖和凋亡的影响。结果显示,与MOCK和DMSO组相比,YM155在0.005μmol/L和0.01μmol/L浓度下均能够显著抑制SU_DIPG13细胞增殖(图3D),促进细胞凋亡(图3E)。
2.4 DIPG原代细胞的靶向小分子组合筛选与验证
为了解决DIPG治疗中HDAC抑制剂的获得性耐药和BET抑制剂未能显著诱导细胞毒杀伤的问题[8],本研究在单药筛选的基础上进行药物库与HDAC抑制剂panobinostat和BET抑制剂JQ1的组合筛选。
在组合筛选中,将66种药物在0.01、0.1、1、10μmol/L浓度下分别与浓度为IC40的panobinostat和JQ1进行组合筛选,并分别计算单药、JQ1组合筛选和panobinostat组合筛选中各个小分子在不同浓度下的肿瘤细胞活性抑制率。然后以同一药物浓度下panobinostat组或JQ1组较DMSO组抑制率高10%作为标准,筛选具有潜在协同抑制作用的小分子组合(图4A)。
为了验证以上组合筛选结果可靠性,在panobinostat组和JQ1组中各挑选了一个未在DIPG中被报道过的组合[HDAC抑制剂与B细胞淋巴瘤/白血病-2基因(B-cell lymphoma/leukemia gene-2,Bcl-2)抑制剂、BET抑制剂与CK2抑制剂]在2个DIPG原代细胞中进行体外组合抑制测试。其中ABT-737为死亡结构关键结构域BH3的模拟抑制剂,作用于Bcl-2[18];CX4945是一种有效的、可口服的ATP竞争性的CK2抑制剂[19]。分别测试4个浓度条件下的单药抑制率和组合抑制率,并计算其CI。结果显示,除了SU_DIPG17细胞中panobinostat与ABT-737的最低浓度组合条件之外,其他组合其CI均小于1,说明HDAC抑制剂与Bcl-2抑制剂以及BET抑制剂与CK2抑制剂之间存在针对DIPG细胞的协同抑制作用(图4B、C)。以上验证结果表明我们筛选发现的一系列具有协同抑制作用的DIPG靶向组合新策略具备进一步进行体内验证的潜力。

图4 DIPG的体外药物组合筛选及验证Fig 4 Screening and validation of combinatory drug for DIPG in vitro
3 讨论
DIPG是一类发病率高且极为恶性的儿童脑肿瘤,目前仅放疗具有一定的效果,因此亟需寻找有效的靶向治疗方法[6]。在本研究中,我们根据DIPG肿瘤组织的转录组分析选择多个靶向小分子在患者来源DIPG原代细胞中进行了体外筛选。转录组分析结果显示,在DIPG中显著上调的基因富集于细胞外基质和细胞表面,参与微管、细胞外基质等的形成和功能;这些在DIPG中表达上调的基因可能通过激活细胞外基质以及核糖体相关的信号通路促进肿瘤细胞增殖[20]。与之一致的是,在DIPG中已经有血小板源性生长因子受体α多肽(platelet-derived growth factor receptorα,PDGFRA)、RAS、1型激活素受体(activin receptor type-1,ACVR1)以及成纤维生长因子受体(fibroblast growth factor receptor,FGFR)等多条细胞外基质或膜表面相关信号通路被发现具有重要致癌作用[21-24]。DIPG中显著下调的基因富集于突触,参与突触和细胞以及细胞之间的信号转导和细胞分化。突触通常随着神经系统的发育而不断形成[25]。DIPG中与神经突触、信号传递功能相关的基因表达显著下调,表明其具有去分化或分化抑制特性,而这种分子特征对于肿瘤的形成和维持可能具有重要作用[26],这与已有研究结果一致[22,27]。
结合筛选和体外验证结果,我们鉴定出DIPG中显著高表达的BIRC5基因的靶向小分子药物YM155对DIPG细胞具有较强的体外杀伤效果。进一步的研究表明YM155能够显著抑制DIPG原代细胞增殖并诱导其凋亡。这为DIPG的靶向治疗提供了一种新策略。YM155已经进入肿瘤治疗的临床试验(表1)。本研究结果为该药物的相关研究成果尽快进行临床转化提供了重要基础。
目前已经明确H3K27M组蛋白突变引起的表观遗传失调在DIPG中发挥了关键的肿瘤驱动功能[28-29],但该突变本身并不是合适的药物靶标。针对这个情况,近年来包括我们在内的多个研究团队开辟了DIPG的靶向治疗新方向,揭示了多个能在临床前肿瘤模型中有效杀伤DIPG的表观遗传靶向策略,包括BET抑制剂、HDAC抑制剂和CDK7抑制剂等[8-9]。其中最值得关注的是HDAC抑制剂panobinostat,其已经进入治疗DIPG的Ⅰ期临床试验。但我们发现药物长期作用可诱导DIPG细胞产生针对panobinostat的获得耐药性,BET抑制剂也存在不能显著诱导细胞毒作用的短板[8]。为了解决这些问题,我们同时进行了基于BET抑制剂或HDAC抑制剂的组合筛选和初步验证。筛选结果中panobinostat与JQ1以及与PR-171(蛋白酶体抑制剂)的协同抑制作用已在DIPG中有过报道[30];而panobinostat与PR-171或RAD001(mTOR抑制剂)的潜在组合已分别进入多发性骨髓瘤(NCT01301807)和淋巴瘤(NCT00967044)的临床试验,这些均说明我们的筛选方法有效。在此基础上发现HDAC抑制剂与Bcl-2抑制剂以及BET抑制剂与CK2抑制剂等新的潜在联合药物治疗策略,为后续进行针对DIPG体内临床前肿瘤模型的组合治疗验证提供了初步证据。
DIPG是发生在脑干中脑桥区域的颅内肿瘤类型,血脑屏障的存在会显著阻碍多种小分子药物进入肿瘤区域。因此与其他脑肿瘤一样,DIPG的药物治疗面临着更大的挑战[31-32]。本研究发现的能在单药或组合治疗中显著抑制DIPG的靶向小分子HDAC抑制剂、BET抑制剂和CK2抑制剂均能有效通过血脑屏障,且已有药物(panobinostat、OTX015、CX4945)进入脑肿瘤治疗的早期临床试验,但Bcl-2抑制剂以及BIRC5抑制剂类药物尚无法有效跨越血脑屏障[33-35]。因此,在后续应用体内模型进行药物治疗测试时,要么选取可有效通过血脑屏障的同类靶向小分子药物,要么通过特殊的方法来实现颅内有效药物递送。对流增强递送(convection enhanced delivery,CED)是一种直接将药物注入肿瘤所在部位从而克服血脑屏障的颅内药物递送技术,目前正在临床上开展脑肿瘤治疗的试验[36-37]。已有研究[38-39]表明该方法同样适用于DIPG的治疗,其与免疫放射疗法联合后表现出良好的安全性和可行性。因此,该方法的应用有望大大促进我们所发现的DIPG靶向新策略的体内验证与临床转化。
综上所述,我们通过转录组分析和靶向小分子单药或组合筛选,发现并初步验证了DIPG的一些潜在有效靶向治疗策略,为它们进一步在DIPG的体内临床前肿瘤模型中的研究提供了初步证据。后续更加深入的研究有望为DIPG这类严重危害儿童健康的疾病提供新的治疗思路。
参·考·文·献
[1] Siegel RL,Miller KD,Fuchs HE,et al.Cancer statistics,2021[J].CA Cancer JClin,2021,71(1):7-33.
[2] Ostrom QT,Cioffi G,Gittleman H,et al.CBTRUS statistical report:primary brain and other central nervous system tumors diagnosed in the United States in 2012—2016[J].Neuro Oncol,2019,21(Suppl 5):v1-v100.
[3] Hoffman LM,Veldhuijzen van Zanten SEM,Colditz N,et al.Clinical,radiologic,pathologic,and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma(DIPG):a collaborative report from the international and European society for pediatric oncology DIPG registries[J].JClin Oncol,2018,36(19):1963-1972.
[4] Aziz-Bose R,Monje M.Diffuse intrinsic pontine glioma:molecular landscape and emerging therapeutic targets[J].Curr Opin Oncol,2019,31(6):522-530.
[5] Zhang X,Zhang Z.Oncohistone mutations in diffuse intrinsic pontine glioma[J].Trends Cancer,2019,5(12):799-808.
[6] Warren KE.Diffuse intrinsic pontine glioma:poised for progress[J].Front Oncol,2012,2:205.
[7] Margueron R,Reinberg D.The polycomb complex PRC2 and its mark in life[J].Nature,2011,469(7330):343-349.
[8] Nagaraja S,Vitanza NA,Woo PJ,et al.Transcriptional dependencies in diffuse intrinsic pontine glioma[J].Cancer Cell,2017,31(5):635-652.e6.
[9] Grasso C,Tang Y,Truffaux N,et al.Functionally-defined therapeutic targets in diffuseintrinsic pontine glioma[J].Nat Med,2015,21(6):555-559.
[10] Borrelli EP,McGladrigan CG.Differences in safety profiles of newly approved medications for multiple myeloma in real-world settingsversusrandomized controlled trials[J]. J Oncol Pharm Pract, 2020:1078155220941937.
[11] Spriano F,Stathis A,Bertoni F.Targeting BET bromodomain proteins in cancer:theexampleof lymphomas[J].Pharmacol Ther,2020,215:107631.
[12] Altieri DC.Survivin,cancer networks and pathway-directed drug discovery[J].Nat Rev Cancer,2008,8(1):61-70.
[13] Kim YH,Kim SM,Kim YK,et al.Evaluation of survivin as a prognostic marker in oral squamous cell carcinoma[J].JOral Pathol Med,2010,39(5):368-375.
[14] Kelly RJ,Thomas A,Rajan A,et al.A phaseⅠ/Ⅱstudy of sepantronium bromide(YM155,survivin suppressor)with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer[J].Ann Oncol,2013,24(10):2601-2606.
[15] Tolcher AW,Quinn DI,Ferrari A,et al.A phaseⅡstudy of YM155,a novel small-molecule suppressor of survivin,in castration-resistant taxanepretreated prostate cancer[J].Ann Oncol,2012,23(4):968-973.
[16] Dahl NA,Danis E,Balakrishnan I,et al.Super elongation complex as a targetable dependency in diffuse midline glioma[J].Cell Rep,2020,31(1):107485.
[17] Chou TC.The combination index(CI<1)as the definition of synergism and of synergy claims[J].Synergy,2018,7:49-50.
[19] Lin GL,Wilson KM,Ceribelli M,et al.Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening[J].Sci Transl Med,2019,11(519):eaaw0064.
[18] Tse C,Shoemaker AR,Adickes J,et al.ABT-263:a potent and orally bioavailable Bcl-2 family inhibitor[J].Cancer Res,2008,68(9):3421-3428.
[20] Siddiqui-Jain A,Drygin D,Streiner N,et al. CX-4945,an orally bioavailable selective inhibitor of protein kinase CK2,inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy[J].Cancer Res,2010,70(24):10288-10298.
[21] Eke I,Cordes N.Focal adhesion signaling and therapy resistance in cancer[J].Semin Cancer Biol,2015,31:65-75.
[22] Schramm K,Iskar M,Statz B,et al.DECIPHER pooled shRNA library screen identifies PP2A and FGFR signaling as potential therapeutic targets for diffuseintrinsic pontine gliomas[J].Neuro Oncol,2019,21(7):867-877.
[23] Fortin J,Tian RX,Zarrabi I,et al.Mutant ACVR1 arrests glial cell differentiation to drive tumorigenesis in pediatric gliomas[J].Cancer Cell,2020,37(3):308-323.e12.
[24] Koncar RF,Dey BR,Stanton AJ,et al.Identification of novel RAS signaling therapeutic vulnerabilities in diffuse intrinsic pontine gliomas[J].Cancer Res,2019,79(16):4026-4041.
[25] Larson JD,Kasper LH,Paugh BS,et al.Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression[J].Cancer Cell,2019,35(1):140-155.e7.
[26] Batool S,Raza H,Zaidi J,et al.Synapse formation:from cellular and molecular mechanisms to neurodevelopmental and neurodegenerative disorders[J].JNeurophysiol,2019,121(4):1381-1397.
[27] Friedmann-Morvinski D,Bushong EA,Ke E,et al.Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice[J].Science,2012,338(6110):1080-1084.
[28] Filbin MG,Tirosh I,Hovestadt V,et al.Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq[J].Science,2018,360(6386):331-335.
[29] Iwagawa T,Watanabe S.Molecular mechanisms of H3K27me3 and H3K4me3 in retinal development[J].Neurosci Res,2019,138:43-48.
[30] Deaton AM,Bird A.CpG islands and the regulation of transcription[J].Genes Dev,2011,25(10):1010-1022.
[31] Lin GL,Wilson KM,Ceribelli M,et al.Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening[J].Sci Transl Med,2019,11(519):eaaw0064.
[32] van Tellingen O,Yetkin-Arik B,de Gooijer MC,et al.Overcoming the blood-brain tumor barrier for effective glioblastoma treatment[J].Drug Resist Updat,2015,19:1-12.
[33] Arvanitis CD,Ferraro GB,Jain RK.The blood-brain barrier and blood-tumour barrier in brain tumoursand metastases[J].Nat Rev Cancer,2020,20(1):26-41.
[34] Minematsu T,Sonoda T,Hashimoto T,et al.Pharmacokinetics,distribution and excretion of YM155 monobromide,a novel small-molecule survivin suppressant,in male and pregnant or lactating female rats[J].Biopharm Drug Dispos,2012,33(3):160-169.
[35] Cheng ZX,Gong YY,Ma YF,et al.Inhibition of BET bromodomain targets genetically diverse glioblastoma[J].Clin Cancer Res,2013,19(7):1748-1759.
[36] Matzuk MM,McKeown MR,Filippakopoulos P,et al.Small-molecule inhibition of BRDT for male contraception[J].Cell,2012,150(4):673-684.
[37] Allard E,Passirani C,Benoit JP. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors[J].Biomaterials,2009,30(12):2302-2318.
[38] Vogelbaum MA,Aghi MK.Convection-enhanced delivery for the treatment of glioblastoma[J].Neuro Oncol,2015,17(Suppl 2):ii3-ii8.
[39] Souweidane MM,Kramer K,Pandit-Taskar N,et al.Convection-enhanced delivery for diffuse intrinsic pontine glioma:a single-centre,dose-escalation,phase 1 trial[J].Lancet Oncol,2018,19(8):1040-1050.
[40] Luther N,Zhou ZP,Zanzonico P,et al.The potential of theragnostic¹²⁴I-8H9 convection-enhanced delivery in diffuse intrinsic pontine glioma[J].Neuro Oncol,2014,16(6):800-806.
猜你喜欢
黑龙江科学(2023年8期)2023-06-04 08:40:58
保健医苑(2022年5期)2022-06-10 07:46:38
中国循证心血管医学杂志(2021年10期)2021-11-05 06:56:00
中国临床医学影像杂志(2021年6期)2021-08-14 02:21:56
肝博士(2020年5期)2021-01-18 02:50:18
解放军医学院学报(2020年12期)2020-03-29 05:11:32
心肺血管病杂志(2019年9期)2019-12-09 08:34:02
中成药(2018年9期)2018-10-09 07:18:32
台州学院学报(2018年6期)2018-02-26 06:14:38
医学研究杂志(2015年7期)2015-06-22 11:01:01
