
IL-37对脂多糖诱导AR42J细胞炎症反应及p38MAPK/NF-κB通路的影响
2021-09-07 02:34:58韩永艳司金春杨林赵国尧刘慧丽陈培莉
广东医学 2021年8期
韩永艳, 司金春, 杨林, 赵国尧, 刘慧丽, 陈培莉△
1商丘市第一人民医院(徐州医科大学商丘临床学院)重症医学科(河南商丘 476100); 2商丘医学高等专科学校临床医学院(河南商丘 476000)
急性胰腺炎属于临床危重疾病,起病迅速,病死率极高可达30%以上[1]。急性胰腺炎发病机制较复杂,目前认为胰腺腺泡中炎症反应是造成发病的重要原因,胰腺炎发生早期阶段,胰腺腺泡细胞释放大量的炎性因子及趋化因子,损伤细胞,且进一步引发全身炎症反应,因此深入探究胰腺腺泡细胞早期炎症反应对于阐明急性胰腺炎发病机制具有重要意义[2]。p38有丝分裂原活化蛋白激酶(MAPK)是调节炎症反应的重要通路,与急性胰腺炎的发生密切相关[3],此外p38MAPK信号通路可与核转录因子 κB(NF-κB) 通过交互作用共同影响炎症的发生[4]。白细胞介素(IL)-37属于IL-1家族成员之一,研究发现重组IL-37蛋白能够降低脂多糖(LPS)引发的炎症反应,然而在急性胰腺炎中的作用,目前研究不多[5]。2016年9月至2019年6月,本研究以胰腺外分泌AR42J细胞为研究对象,转染IL-37过表达载体后给予LPS诱导细胞炎症反应,旨在探究IL-37对AR42J细胞炎症反应的影响及可能的作用机制。
1 材料与方法
1.1 实验材料 大鼠胰腺外分泌AR42J细胞购于中国科学院细胞生物研究所;采取含10%血清的RPMI-1640培养基培养细胞。
1.2 主要试剂与仪器 胎牛血清及RPMI-1640培养基购于美国Gibco公司,货号:10270-106,1800-022;pcDNA3.1质粒购于上海柯雷生物科技有限公司公司,货号:kl-zl-0650;CCK-8细胞增殖检测试剂盒购于上海生工生物工程公司,货号:B518229;脂质体转染试剂盒购于美国Invitrogen公司,货号:11668-017;BCA检测试剂盒、RIPA裂解液购于碧云天生物技术有限公司;TNF-α、IL-6、IL-1β检测试剂盒购于南京建成生物工程研究所,货号:H054,H007,H002;MAPK、p38MAPK抗体、IL-37抗体购于英国Abcam公司,货号:ab227426,ab4822,ab101376;NF-κB、LaminB1、β-actin抗体购于公司,货号SAB4501993,CLL1038,F3022:FC酶标仪购于美国Thermo公司;3412Transwell小室购于康宁生物器材有限公司;DSZ-70PHC光学显微镜购于日本Carton公司;GelDocXR凝胶成像仪购于美国BIO-RAD公司。
1.3 实验方法
1.3.1 细胞转染与实验分组 采取含10% FBS的RPMI培养基在37℃ 5% CO2潮湿培养箱内培养AR42J细胞, 传代培养至第3代后,收集对数生长期细胞接种6孔细胞培养板中,添加不含FBS的培养液培养1 h后进行转染,具体参照脂质体转染试剂盒说明书进行操作,将细胞分为3组:(1)空白组:细胞不进行任何处理,正常培养;(2)阴性对照组:将pcDNA3.1质粒转染至AR42J细胞;(3)IL-37过表达组:将pcDNA3.1-IL-37质粒转染至AR42J细胞。以上3组细胞转染后,在37℃ 5% CO2潮湿培养箱继续培养6 h,更换为含FBS的培养液,培养液中添加10 μg/mL LPS,常规培养8 h收集上述各组细胞。
1.3.2 CCK8法检测AR42J细胞增殖活性 收集各组细胞,添加胰酶消化后制备单细胞悬液,细胞密度调整为2×104个/mL,接种100 μL至细胞培养板内,每组设置3个重复,常规培养当细胞融合度在70%时,添加10 μL CCK8检测液,继续孵育4 h后,采用酶标仪检测各孔450 nm波长处光密度值(OD),并计算隔空细胞增殖率=待测组OD/空白组OD×100%,每组均重复检测3次。
1.3.3 酶联免疫吸附法检测各组细胞中TNF-α、IL-6、IL-1β水平 采取酶联免疫检测法检测细胞中TNF-α、IL-6、IL-1β水平,具体严格按照试剂盒说明书严格操作。
1.3.4 AR42J细胞淀粉酶分泌率检测 采取酶动力学法检测细胞培养液及细胞中淀粉酶酶活,计算淀粉酶分泌率,淀粉酶分泌率=细胞培养液中淀粉酶/细胞中淀粉酶×100%。
1.3.5 免疫印迹法检测细胞中p38MAPK/NF-κB蛋白表达情况 收集各组细胞,加入细胞裂解液RIPA提取细胞蛋白,以BCA法检测细胞总蛋白,统一定量30 μg蛋白,经SDS-PAGE电泳分离蛋白后,移至PVDF膜湿转法转膜后,经脱脂奶粉室温下封闭膜后,用p-p38MAPK、p38MAPK、IL-37、NF-κB、LaminB1、β-actin一抗抗体(1∶300)4℃下过夜孵育膜,洗膜后用二抗抗体(1∶5 000)室温下温育膜2 h,洗膜后,经ECL发光剂曝光图片后,采用凝胶成像扫描仪扫描蛋白条带,以β-actin作为内参,用Image-J软件定量分析条带相对表达量。

2 结果
2.1 转染后细胞中IL-37蛋白表达情况 与空白组与阴性对照组相比, IL-37过表达组AR42J细胞中IL-37蛋白表达量显著升高(P<0.05)。见图1和表1。
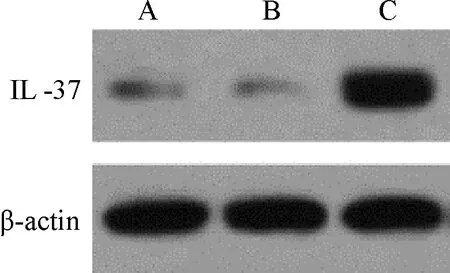
注:A:空白组;B:阴性对照组;C: IL-37过表达组
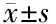
表1 转染后细胞中IL-37蛋白表达情况
2.2 IL-37过表达对AR42J细胞增殖活性的影响 随着AR42J细胞培养时间的延长,与空白组与阴性对照组相比, IL-37过表达组AR42J细胞增殖率显著升高(P<0.05)。见表2。
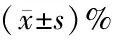
表2 各组AR42J细胞增殖抑制率比较(n=6)
2.3 IL-37过表达对AR42J细胞炎症因子的影响 与空白组与阴性对照组相比, IL-37过表达组AR42J细胞TNF-α、IL-1β、IL-6水平显著降低(P<0.05)。见表3。
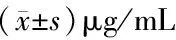
表3 各组AR42J细胞TNF-α、IL-1β、IL-6水平比较(n=6)
2.4 IL-37过表达对淀粉酶活性的影响 与空白组与阴性对照组相比, IL-37过表达组AR42J细胞淀粉酶分泌率显著降低(P<0.05)。见表4。

表4 各组AR42J细胞淀粉酶活比较(n=6)
2.5 IL-37过表达对AR42J细胞p38MAPK蛋白表达的影响 与空白组与阴性对照组相比, IL-37过表达组AR42J细胞p38MAPK蛋白磷酸化水平显著降低(P<0.05)。见图2和表5。

注:A:空白组;B:阴性对照组;C: IL-37过表达组
表5 各组AR42J细胞p-p38MAPK、p38MAPK蛋白表达比较(n=6)
2.6 IL-37过表达对AR42J细胞NF-κB蛋白表达的影响 与空白组与阴性对照组相比, IL-37过表达组AR42J细胞核NF-κB蛋白表达显著降低(P<0.05),细胞质NF-κB蛋白表达水平比较差异无统计学意义(P>0.05)。见图3和表6。
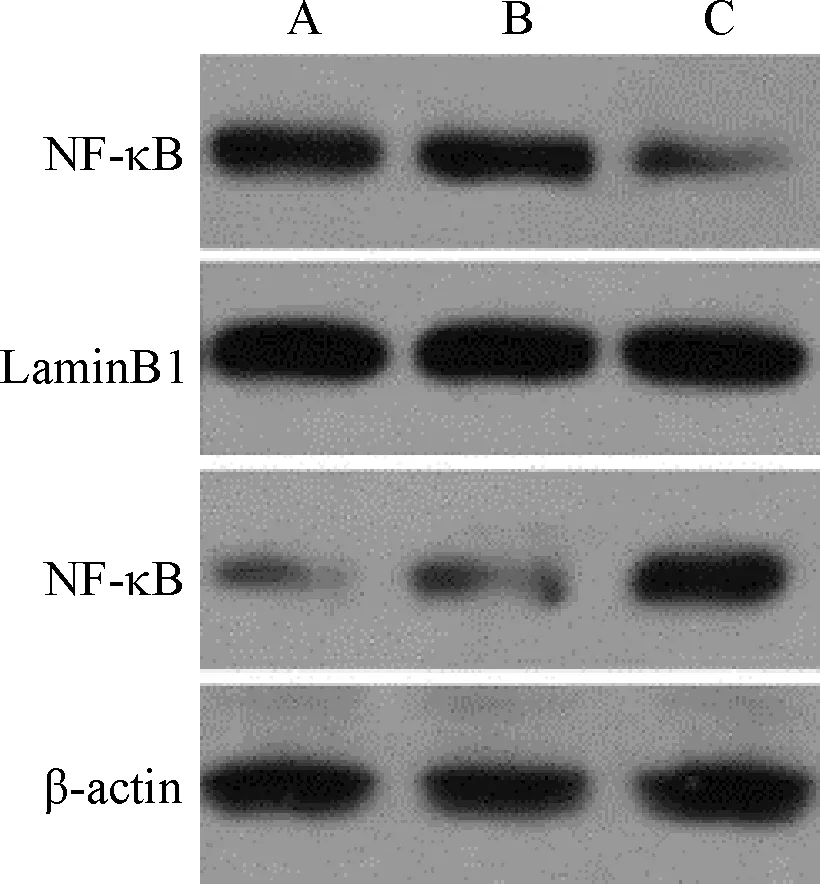
注:A:空白组;B:阴性对照组;C: IL-37过表达组
表6 各组AR42J细胞NF-κB蛋白表达比较(n=6)
3 讨论
AR42J细胞属于大鼠胰腺腺泡细胞,与人体正常胰腺腺泡细胞功能相似,能够分泌、合成消化酶,进行信号传导等,多用在胰腺体体外实验[6]。LPS属于革兰阴性菌细胞壁的主要成分,研究证实LPS参与急性胰腺炎的发病过程[7]。LPS可使轻度胰腺炎发展为重症胰腺炎,进一步造成系统炎症综合征的发生[8]。研究发现LPS能够诱导刺激AR42J细胞产生急性炎症反应,且重复性较高,是研究胰腺炎良好的体外细胞模型,因此本研究也采用LPS刺激AR42J细胞复制胰腺炎细胞模型[9]。IL-37属于抗炎因子,在胸腺、淋巴结等器官中均正常表达。在正常状态下,IL-37呈低表达,当受到炎症反应刺激后,其表达明显升高,可抑制炎症反应。本研究通过将IL-37过表达载体转染至AR42J细胞中孵育后给予LPS刺激,结果发现与空白组、阴性对照组相比,细胞所释放的炎症因子如TNF-α、IL-1β、IL-6水平显著降低,空白组与阴性对照组因子水平无明显变化,表明IL-37能够抑制LPS诱发的炎症因子,进而降低细胞炎症反应。
LPS能够刺激巨噬细胞分泌大量炎症因子进而损伤腺泡细胞,有研究发现小剂量的LPS能够诱发腺泡细胞凋亡,而高剂量的LPS能够造成细胞水肿,使胞质膜、溶酶体破坏,进而导致淀粉酶分泌升高[10-11]。本研究发现与空白组和阴性对照组相比,IL-37过表达组细胞增殖活力升高,淀粉酶分泌率降低,推测IL-37可通过降低胰腺腺泡细胞炎症反应进而改善细胞活性,抑制淀粉酶过量分泌。
MAPK信号转导通路广泛存在细胞中,主要参与细胞增殖、发育及胞间信号传递过程。MAPK通路主要包括p38 MAPK、ERK、JNK三条通路,这3条信号通路可独立或相互交联发挥作用[12]。其中p38MAPK通路与炎症反应调节机制有关,在未受到炎症刺激时呈低表达,当受到炎症因子刺激后磷酸化水平升高,进而调节各类炎性因子表达[13-14]。研究发现在急性胰腺炎大鼠中,p38MAPK磷酸化水平明显升高,可促进炎症因子IL-8、TNF-α大量释放,造成炎症反应失调,最终引发级联反应[15]。此外研究发现采用p38 MAPK通路抑制剂处理急性胰腺炎大鼠,发现能够降低机体炎症因子水平,降低胰腺组织病理损伤[16]。这些研究均表明p38MAPK活化与胰腺炎的发生密切相关。本研究发现与空白组、阴性对照组相比,IL-37过表达组细胞中p-p38MAPK蛋白表达显著升高,推测p38MAPK信号通路参与LPS诱导腺泡细胞炎症反应过程,IL-37可通过抑制p38MAPK通路活化,进而抑制腺泡细胞AR42J炎症反应。
NF-κB广泛参与炎症反应及免疫反应,近期发现NF-κB参与胰腺炎的病理发生过程,Pu等[17]研究发现黄芩素还能抑制LPS激活的腺泡细胞AR42J炎症反应,抑制NF-κB的激活,进而降低炎症因子水平来抑制导管细胞化生。Pan等[18]研究发现LPS刺激腺泡细胞AR42J后可激活NF-κB通路,抑制细胞增殖,诱导细胞凋亡,而抑制NF-κB通路表达后可部分逆转LPS刺激的作用。本研究发现与空白组、阴性对照组相比,IL-37过表达组细胞核NF-κB蛋白表达显著降低,推测NF-κB信号通路参与LPS诱导腺泡细胞炎症反应,IL-37可通过抑制NF-κB通路活化,进而抑制腺泡细胞AR42J炎症反应。
研究发现在炎症反应过程中NF-κB可受到p38MAPK信号通路激活,释放大量炎症因子进而造成级联反应,为局部炎症向全身发展提供了条件[19]。Kumar等[20]研究显示可在巨噬细胞中下调NF-κB和MAPK信号通路,进而降低TNF-α、IL-6、MCP-1和IL-1β的表达,降低巨噬细胞炎症反应。基于此本研究推测IL-37可能通过抑制胰腺腺泡AR42J细胞p38MAPK信号通路,进而抑制NF-κB通路活化,下调细胞炎症因子,降低细胞炎症反应。
综上所述,IL-37能够抑制LPS诱导AR42J细胞炎症反应,其机制可能与抑制胰腺腺泡AR42J细胞p38MAPK信号通路,进而抑制NF-κB通路活化,下调细胞炎症因子有关,IL-37的这一发现为胰腺炎治疗提供新的思路,然而本研究也存在一定的不足之处,IL-37具体通过NF-κB下游哪些因子发挥抗炎作用,仍待后续深入研究。
猜你喜欢
中华胰腺病杂志(2023年5期)2023-10-27 07:32:10
临床与实验病理学杂志(2022年11期)2022-02-15 16:20:11
中国生殖健康(2019年2期)2019-08-23 08:11:58
中国医学影像学杂志(2018年9期)2018-10-17 01:26:58
中国医药指南(2017年3期)2017-11-13 02:55:17
中华胰腺病杂志(2015年5期)2015-12-08 12:18:13
医学研究杂志(2015年12期)2015-06-10 06:57:46
中国药理学通报(2014年2期)2014-05-09 08:22:30
中医研究(2014年5期)2014-03-11 20:28:49
中医研究(2013年5期)2013-03-11 20:26:55
