
转录因子BTB 与CNC 同源蛋白2 在血液恶性肿瘤中的研究进展△
2021-09-04 06:21王佳张寒
癌症进展 2021年10期
王佳,张寒
1中国医学科学院医学生物学研究所,昆明 650118
2云南大学生命科学学院,昆明 650500
淋巴瘤是起源于淋巴造血系统的恶性肿瘤,而白血病是一类造血干细胞恶性克隆性疾病。肿瘤细胞由于分化障碍、增殖失控、凋亡受阻等机制在骨髓及造血组织中大量增殖,从而引发严重的临床症状与并发症。随着多药联合化疗与支持疗法的应用,淋巴瘤与白血病的治愈率有明显提升。尽管如此,二者的发病机制与复发机制至今仍不明确,且很多亚型缺乏特异性分子靶标。例如,儿童T 细胞急性淋巴细胞白血病(acute lymphoblastic leukemia,ALL)在临床易复发,且目前缺乏较为理想的治疗靶标。此外,大量研究相继报道了抗淋巴瘤药物如硼替佐米(bortezomib,BTZ)与依鲁替尼的耐药反应,但关于耐药机制尚无定论。因此,迫切需要发现新的分子靶标,为阐明血液肿瘤发病机制及个性化治疗提供新思路。
BTB 与CNC 同 源 蛋 白2(BTB domain and CNC homolog 2,BACH2)是B 淋巴细胞特异性转录因子,由BACH2
基因编码,在B 细胞定向发育过程中发挥关键作用,并参与多种先天性与适应性免疫反应。在淋巴祖细胞阶段,BACH2 抑制髓系发育程序,推动祖细胞向B 细胞分化。在前体B(pre-B)细胞阶段,BACH2 与B 细胞淋巴瘤/白血病-6(B cell lymphoma/leukemia-6,Bcl-6)竞争性调控基因的启动子区,对B 细胞进行阴性选择。在B 细胞成熟阶段,BACH2 与Bcl-6 进一步协作阻止B 细胞向浆细胞分化,并调节抗体类别转换重组与体细胞高频突变。除了B 细胞,BACH2 还参与T细胞介导的免疫反应及T 细胞静息状态的维持。以往研究多关注BACH2 在人体免疫自稳调节及炎性反应中的作用,然而最近研究发现BACH2 在血液恶性肿瘤中发挥重要的生物学功能。据此,本文将重点围绕BACH2 的结构与功能在血液恶性肿瘤中的研究情况进行综述。1 BACH2 蛋白结构与功能
BACH 蛋白家族是碱性亮氨酸拉链(basic region-leucine zipper,bZIP)转录因子蛋白家族成员之一,由BACH1 与BACH2 组成。其中,BACH1 广泛存在于各种组织中,而BACH2主要在T/B细胞中特异性表达。除了BACH家族外,人类bZIP转录因子还包括很多其他成员,例如激活蛋白-1(activator protein-1,AP-1)、核因子红细胞2(nuclear factorerythroid 2,NF-E2)、NF-E2 相关因子2(NF-E2-related factor 2,NRF2)及MAF 蛋白等。在结构上,BACH2 主要由BTB 与bZIP 结构域组成(图1A)。
1.1 BTB 结构域
BTB 结构域位于BACH2 蛋白的氨基(N)末端,核心结构包括5 个α-螺旋(A1~A5)与3 个β-折叠(B1~B3)。其中,A1/A2 与A4/A5 分别形成两个α-螺旋发卡结构,而B1/B2/B3 形成一个β片层结构。在此基础上,B1/B2/A1/A2/B3 区域通过A3 和一个可变连接区域连接A4/A5 区域(图1B)。除了核心结构外,不同亚型的BTB 还包括N 末端与羧基(C)末端的延伸区域,该区域赋予蛋白某些特定功能。

图1 BACH2结构示意图
BTB 结构域主要参与蛋白的同源或异源二聚化与多聚化反应,并辅助参与抑制因子复合物的蛋白互作。例如,BACH2 通过与组蛋白去乙酰化酶3(histone deacetylase 3,HDAC3)相互作用抑制PR/SET 结构域1(PR/SET domain 1,PRDM1)基因的转录,从而阻止浆细胞的终末分化。
1.2 bZIP 结构域
bZIP 结构域主要由DNA 结合结构域与亮氨酸拉链(leucine zipper,ZIP)结构域组成。DNA 结合结构域也称作碱性结构域(basic domain),可与DNA 序列上的TPA 反应元件(TPA response element,TRE)或cAMP 反应元件(cAMP response element,CRE)特异性结合。由于富含高度保守的碱性氨基酸,DNA 结合结构域可稳定蛋白质与DNA 之间的相互作用。
ZIP 结构域是一个由60~80 个氨基酸组成的七肽重复序列,每个该结构域的第7 位上都是亮氨酸,且每个七肽重复序列跨越两个α螺旋。其中,第1 个七肽重复序列通过一个短铰链区与DNA 结合结构域相连,组成bZIP 单体。ZIP 结构域的保守性较差,并与其他bZIP 因子的ZIP 结构域形成二聚卷曲螺旋结构,该结构使得两个bZIP 单体在DNA周边组装成同源或异源二聚体,并通过碱性区域分别与DNA 互补链上的回文序列相结合。
二聚反应对于bZIP 转录因子的调控功能具有十分重要的作用。一方面,二聚化使得bZIP 转录因子在DNA 周边形成不同的同源或异源二聚体,发挥不同的调控作用,而与哪一个bZIP 单体发生二聚化主要取决于单体的浓度。另一方面,不同的异源二聚体具有不同的DNA 序列特异性,该特点使得bZIP 单体针对不同的DNA 序列组装成不同的二聚体。一旦bZIP 单体的浓度与结构发生变化,二聚反应也随之调整,并迅速组装成完全不同的二聚体。bZIP 家族的二聚特性还可促进bZIP 单体与抑制或激活因子发生二聚反应。
1.3 BACH2 蛋白的特有结构
不同于其他淋巴转录因子,BACH2 的蛋白活性受到血红素(heme)水平的严格调控。BACH2 包含5 个与血红素结合的半胱氨酸-脯氨酸(cysteineproline,CP)基序,其中3 个CP 位于BACH2 的内在无序区(intrinsically disordered region,IDR)(图1A)。在B 细胞中,BACH2 与血红素结合可抑制BACH2 与DNA 的结合能力,并诱导BACH2 蛋白经蛋白酶体途径发生降解。进一步研究表明,血红素与位于IDR 的CP 基序结合时能诱导BACH2形成非常紧密的构象。这种细微的结构变化足以影响BACH2 的蛋白-蛋白和(或)结构域-结构域互作,从而抑制DNA 结合或蛋白质降解。尽管如此,CP 突变并不能阻止BACH2-血红素互作,提示其他氨基酸残基也可能参与其中。
此外,BACH2 的C 末端还包含一个保守结构,称为细胞质定位信号(cytoplasmic localization signal,CLS)(图1A)。CLS 可介导BACH2 从细胞核向细胞质的输出,去除含有CLS 的BACH2 蛋白C末端可导致BACH2 在胞核内大量累积无法输出,在氧化应激条件下,CLS 的活性被抑制,亦可导致BACH2 核输出障碍。然而,其他蛋白均不包含此结构,提示CLS 是BACH 家族的特有结构。
1.4 BACH2 蛋白的生物学功能
BACH2 蛋白通常与小MAF 蛋白形成异源二聚体,并通过识别DNA 启动子上的TRE 或CRE 发挥转录抑制作用。此外,BACH2 还与其他bZIP因子发生二聚反应,例如,BACH2 与bZIP 转录因子ATF 样蛋白(bZIP transcription factor ATF-like,BATF)形成二聚复合物,并通过干扰BATF-干扰素调节因子4(interferon regulatory factor 4,IRF4)复合物的募集抑制AP
-1
的转录,从而控制辅助性T细胞2(helper T cell 2,Th2)的发育,并在Th2 型肺部炎症中发挥关键作用。由于BACH2 识别的TRE 也被其他bZIP 转录因子所识别,因此二者存在竞争关系,例如,在CD8T 细 胞 中,BACH2 通 过 占 据 多 个AP-1 的DNA 结合位点阻止T 细胞受体(T cell receptor,TCR)驱动的终末效应分化基因的表达。因此,BACH2 与AP-1 之间形成的抑制-激活关系对于BACH2 介导的TCR 驱动的转录抑制程序至关重要。再比如,BACH2 在调节性T 细胞(regulatory T,Treg)谱 系 定 向 后 与 静 息Treg(resting Treg,rTreg)细胞分化基因的增强子结合,并通过与AP-1竞争性结合DNA 抑制TCR 驱动诱导的活化型Treg(activated Treg,aTreg)细胞的分化,从而促进rTreg 细胞静息状态的长期维持。这一功能是维持免疫稳态和持久肿瘤免疫抑制所必需的,可有效避免致死性炎症反应。
此外,BACH2 在免疫球蛋白(immunoglobulin,Ig)基因类别转换重组和体细胞高频突变中也是必不可少的。BACH2 通过转录抑制PRDM1
阻止B 细胞向浆细胞分化,从而赢得活化诱导胞嘧啶核苷脱氨酶(activation-induced cytidine deaminase,AID)蛋白表达的时间窗,以确保抗体类别转换重组与体细胞高频突变的有效完成。尽管如此,小鼠模型中BACH2
缺失的自身反应性B 细胞可以逃脱生发中心(germinal center,GC)检查点,并在滤泡外与BACH2
缺失的Th 细胞协同作用产生IgG 类别转换重组的致病性自身抗体,由此阐明了系统性红斑狼疮伴体液自身免疫的发生机制。除了参与T/B 细胞分化与发育外,细胞质内的BACH2在氧化应激条件下迅速转位至细胞核内抑制抗氧化因子的转录活性,从而促进细胞凋亡。此外,BACH2通过调节抗凋亡及细胞周期相关基因的表达促进B细胞受体诱导的B细胞增殖与存活。
2 BACH2 与淋巴瘤
根据病理学特点,淋巴瘤主要分为霍奇金淋巴瘤与非霍奇金淋巴瘤(non-Hodgkin lymphoma,NHL)。其中,约95%的NHL 为B 细胞来源,又称B 细胞NHL(B-cell NHL,B-NHL),多起源于淋巴组织的GC。B-NHL 又进一步分为弥漫性大B 细胞淋巴瘤(diffuse large B cell lymphoma,DLBCL)、滤泡性淋巴瘤(follicular lymphoma,FL)、伯基特淋巴 瘤(Burkitt lymphoma,BL)及 套 细 胞 淋 巴 瘤(mantle cell lymphoma,MCL)等。
2.1 BACH2 与DLBCL
DLBCL是最常见的B-NHL,约占NHL的30%~40%。早年研究报道,BACH2表达下调提示DLBCL患者预后不良,且BACH2 表达水平是预测DLBCL患者生存率的有效指标。在EB 病毒阳性(epstein-barr virus,EBV)的DLBCL 病例中,BACH2表达下调有助于EBV 存活,并通过转化生长因子-β激活激酶1(transforming growth factor-β-activated kinase 1,TAK1)磷酸化持续性激活核因子κB(nuclear factor-κB,NF-κB)通路,表明BACH2 很可能具有抑制DLBCL 发生发展的功能。
研究发现,TBL1X 受体1(TBL1X receptor 1,TBL1XR1)基因突变通过驱动BACH2 介导的调控产生异常的未成熟记忆B 细胞(memory B cell,MB)。在分子层面,TBL1XR1
突变体迫使核受体辅助抑制因子2(nuclear receptor corepressor 2,NCOR2,又称SMRT)/HDAC3 抑制复合物与Bcl-6发生解离,转而异常结合BACH2,导致前MB 转录重编程及细胞命运的改变。抗原回收后,TBL1XR1
突变型MB 无法正常分化为浆细胞,而是重新进入GC 反应并诱发结外免疫母细胞样淋巴瘤表型。该表型与人活化B 细胞DLBCL 极为相似,该研究为淋巴再循环成瘤提供了有力证据,并从侧面反映了BACH2-Bcl-6转换在B细胞分化中的关键作用。2.2 BACH2 与MCL
MCL 是一种罕见的侵袭性B-NHL,约占NHL的6%。MCL 具有体内多脏器侵犯、对常规化疗不敏感、复发率高、中位生存期短、死亡率高等临床特征,因此被划为难治性淋巴瘤范畴。MCL具有典型的t(11;14)染色体易位,导致细胞周期蛋白D1(cyclin D1,CCND1)持续性高表达,从而促进MCL 进展。然而,约有2%的健康人存在t(11;14)染色体易位,且临床上某些MCL 患者并无t(11;14)染色体易位与CCND1 过表达。在小鼠模型中,过表达CCND1 也不足以诱发肿瘤。这些数据提示MCL 的发生、发展还受到其他分子事件或异常信号转导通路的影响。
研究发现,在MCL 细胞中沉默BACH2
可促进MCL 细胞增殖、加速细胞周期,并显著增加肿瘤细胞的黏附与播散能力;在小鼠模型中,敲除BACH2
不仅加快皮下成瘤,而且增加骨髓及脾脏浸润;在MCL 患者中,BACH2
表达量明显低于正常对照组,且BACH2
下调提示患者预后不良,表明BACH2 在MCL 也发挥类似抑癌因子的作用。然而,在MCL 患者及小鼠骨髓的低氧环境中,BACH2 低表达是由于缺氧诱导因子-1α(hypoxia-induced factor 1α,HIF-1α)的转录抑制与血红素介导的蛋白降解所致。在常氧状态下,BACH2 通过抑制脯氨酰羟化酶3(prolyl hydroxylase 3,PHD3)延缓HIF-1α降解,提示在不同生理条件下,BACH2 与HIF-1α之间存在微妙的调控关系。此外,敲除BACH2
可增加肿瘤细胞对BTZ 和依鲁替尼等药物的耐药反应,部分原因是由于BACH2 低表达促进了恶性B 细胞向浆细胞分化所致。此外,另一项研究发现在BTZ 敏感的MCL 细胞株中,BTZ 诱导产生的活性氧(reactive oxygen species,ROS)使BACH2 迅速转位至细胞核,并通过转录抑制血红素加氧酶-1(heme oxygenase-1,HMOX1)、MCL1
凋亡调节因子等抗氧化及抗凋亡基因诱导细胞发生凋亡。相反,在BTZ 耐药株中,BACH2 则大量滞留在细胞质中无法发挥作用,表明BACH2 的亚细胞定位也是MCL 细胞产生耐药反应的机制之一。(图2)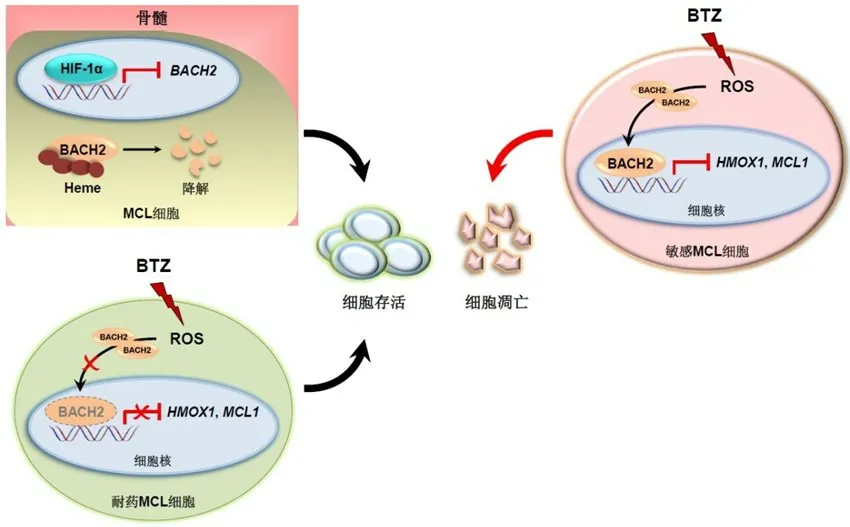
图2 BACH2在MCL中的功能
2.3 BACH2 与其他淋巴瘤
在BL 细胞系中,过表达BACH2 可明显抑制肿瘤细胞增殖并增加化疗药物的毒性反应,提示BACH2 在BL 中也具有抗肿瘤特性。在小鼠模型中,NF-κB 亚单位REL 原癌基因(REL proto-oncogene,c-Rel)缺失可导致小鼠发生早期淋巴瘤;进一步微阵列基因表达分析发现,c
-Rel
缺失可导致BACH2
显著下调。然而,RELA 原癌基因(RELA proto-oncogene,RELA)突变小鼠中的BACH2
表达也下调,且BACH2
低表达的野生型小鼠表现出与c
-Rel
缺失小鼠一样的淋巴瘤早期发病症状。随后研究证实BACH2
在人B 细胞中是c-Rel 的下游靶基因,提示c-Rel 通过转录调节BACH2
在淋巴瘤中发挥抑癌功效。3 BACH2 与白血病
白血病是儿童时期最常见的恶性肿瘤和主要死亡原因之一。中国儿童白血病的发病率为(4~6)/10 万,其中90%以上病例为急性起病,以儿童ALL 最为常见,约占75%以上;慢性白血病起病较隐匿,病程进展缓慢,临床上以慢性粒细胞白血病(chronic myeloid leukemia,CML)和慢性淋巴细胞白血病(chronic lymphocytic leukemia,CLL)最为常见。
3.1 BACH2 与ALL
在正常pre-B 细胞中,BACH2 通过与Bcl-6 竞争性调控下游基因参与B 细胞的阴性选择。当pre-B 细胞中某些癌基因(例如MYC
原癌基因)被激活时,BACH2 通过激活肿瘤蛋白p53(tumor protein p53,TP53)基因诱导细胞凋亡,从而抑制促癌事件的发生;一旦BACH2 表达或活性出现异常,Bcl-6的调控则占据上风,并通过转录抑制TP53
破坏体内的抗癌壁垒,最终导致pre-B 细胞恶性转化。此外,BACH2 的上游调控因子成对盒因子5(paired box 5,PAX5)在pre-B-ALL 细胞中经常发生遗传缺失或功能失活,由此导致的BACH2 下调也是B-ALL 发生的重要因素之一(图3A)。此外,IKAROS 家族锌指蛋白1(IKAROS family zinc finger 1,IKZF1)基因缺失与B-ALL 患者中的BACH2-Bcl-6 轴平衡密切相关。IKZF1
编码的肿瘤抑制因子IKAROS 在淋巴细胞早期发育中发挥关键的调控作用。研究发现,IKAROS 在抑制Bcl
-6
启动子的同时促进BACH2
的转录,而酪蛋白激酶2 抑制剂(CX-4945)正是通过增强IKAROS 功能参与调控Bcl-6-BACH2 轴,诱导B-ALL 细胞发生凋亡。尽管如此,采用CX-4945 对B-ALL 小鼠模型进行单药处理的效果十分有限,且药物处理后的BACH2 与Bcl-6 表达均下调,提示CX-4945 在B-ALL 中的作用机制尚需进一步确认。然而,另一项研究发现组成AP-1 异二聚体的FOS
原癌基因是BACH2 的下游靶点。BACH2 通过负向调控FOS 参与骨髓微环境改变及白血病细胞对阿糖胞苷的耐药反应(图3B),为研究BACH2 在B-ALL 发生、发展过程中的功能提供了新的依据。3.2 BACH2 与CML
CML 的分子特征主要表现为9 号与22 号染色体发生易位,形成费城染色体(Philadelphia chromosome,Ph),该易位使得9 号染色体上的ABL 原癌基因1(ABL proto-oncogene 1,ABL1)与22 号染色体上的BCR
基因形成BCR
-ABL1
融合基因,并编码具有酪氨酸激酶活性的BCR-ABL1 蛋白。因此,酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI)是治疗CML 的主要药物。2001 年,人们首次发现BACH2 表达水平在PhCML 患者中较健康人明显降低;在小鼠模型中,采用TKI 抑制BCR-ABL1 活性可上调BACH2
表达,提示BACH2 很可能受到BCR-ABL1 激酶的转录调控。随后研究发现,PhCML 细胞中的BCR-ABL1 蛋白在氧化应激条件下通过磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)通路介导BACH2 磷酸化,使其滞留在细胞质内无法发生核转位,由此解除了BACH2 对下游靶基因HMOX1
的转录抑制,最终导致CML 恶性转化与存活。此外,BCR-ABL1 蛋白亦可通过PAX5 间接抑制BACH2
转录(图3B),从而促进PhCML 的发生发展。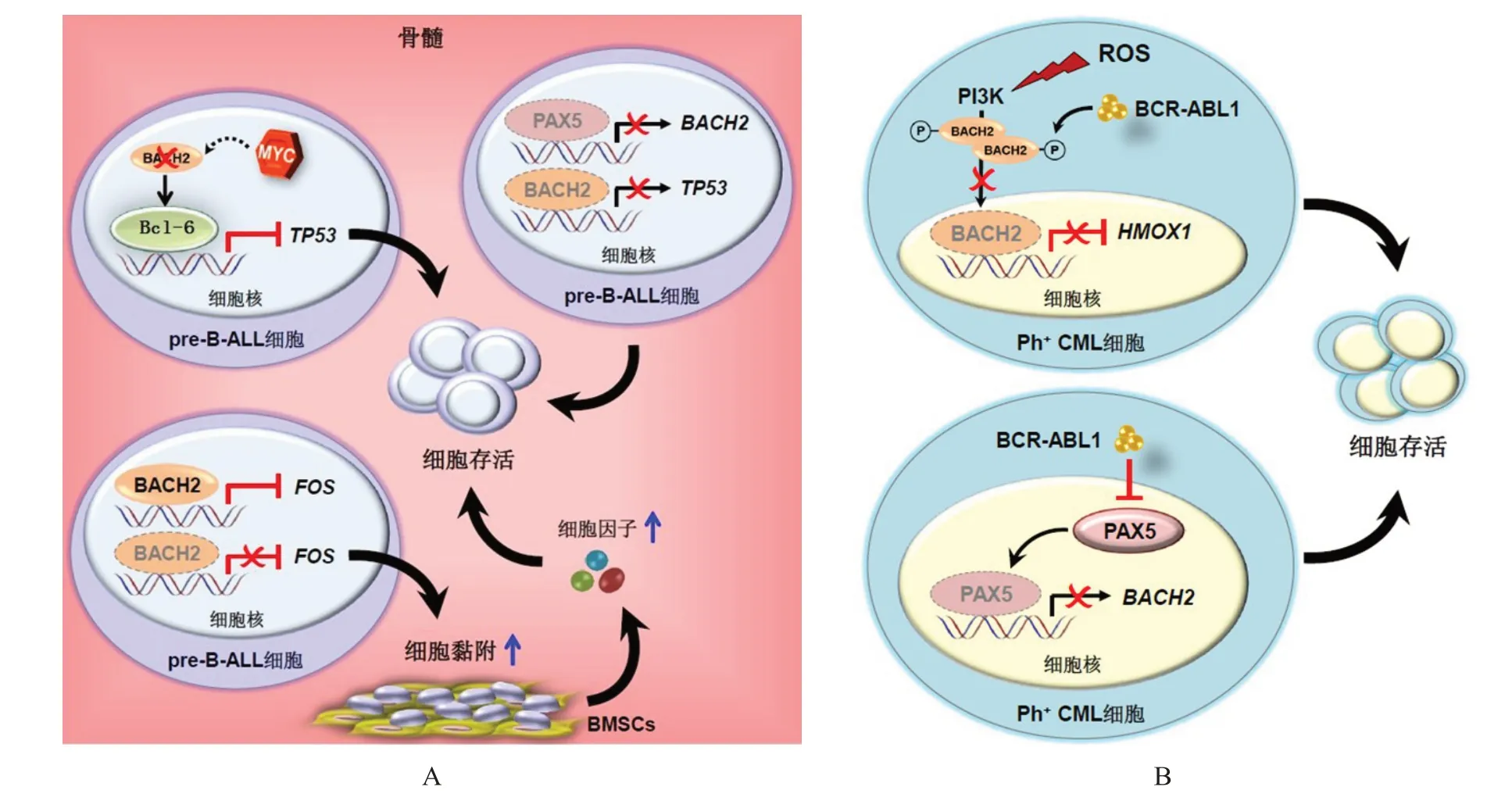
图3 BACH2在ALL中的功能
3.3 BACH2 与CLL
BACH2 在CLL 中的研究鲜有报道,目前仅有一项研究显示BACH2 在CLL 初诊患者的CD4T细胞、CD8T 细胞及CD19B 细胞中较健康人明显降低。值得注意的是,在健康人群的T/B 细胞中,BACH2 的表达水平随着年龄增大逐渐下调,并呈现出明显的抗凋亡能力,提示BACH2 很可能与免疫衰老密切相关。
4 小结与展望
随着人们对BACH2 生理功能的深入探究,BACH2 在淋巴细胞分化及调节中的神秘面纱被慢慢揭开。在B 细胞中,BACH2 通过抑制浆细胞分化促进抗体类别转换重组、体细胞高频突变及MB分化。此外,BACH2 通过调控T 细胞发育及分化维持机体免疫稳态与肿瘤免疫抑制。BACH2 与AP-1 之间的竞争性调控还参与多种关键基因的表达。正因如此,BACH2 功能异常极易诱发血液系统恶性疾病。BACH2 在淋巴瘤或白血病中发挥类似抗肿瘤因子的作用,并参与药物的耐药反应与疾病复发,提示BACH2 很可能成为新的诊断与治疗靶点。尽管如此,针对BACH2研发靶向药物仍存在很大挑战。首先,研制抑癌基因激活剂并使其恢复野生型功能存在一定的技术难点,所以目前仅有两种针对TP53
抗癌基因研发的激活剂获批进入临床试验。其次,在淋巴细胞分化过程中,BACH2 调控的下游网络庞大且复杂。研究发现,BACH2 除了发挥转录调控功能外,还可通过影响染色质重要区域的开放程度参与表观遗传学调控。因此,BACH2 并不是一个十分理想的药物靶点。在未来的研究中,迫切需要关注BACH2 的下游网络并寻找可能的效应因子作为治疗靶标。然而,另有研究发现BACH 家族中的另一个成员BACH1 在实体瘤中发挥促癌功效。BACH1 通过增加糖酵解促进肺癌的转移,且BACH1 高表达与肺癌患者的预后不良密切相关。此外,BACH1 在三阴性乳腺癌患者的肿瘤组织中明显高表达并参与调节线粒体代谢。BACH1 还参与调控干细胞的自我更新。这些研究表明,BACH1在实体瘤中发挥着与BACH2 截然相反的功能。那么在血液肿瘤中,BACH1 与BACH2 之间是否存在相互作用;在氧化应激条件下,BACH1 与BACH2发挥同样的抗氧化调节作用,那么二者在细胞中是如何进行角色分工的;最后,淋巴瘤及白血病患者中的BACH2 表达明显下调,那么BACH2 的功能丧失可否被BACH1 代偿调节;二者在血液肿瘤中又具有怎样不为人知的非经典功能。上述问题不仅指明了未来的研究方向,也为血液恶性肿瘤的个体化和靶向治疗提出了新视角与新思路。
猜你喜欢
新医学(2022年4期)2022-04-23
清华金融评论(2022年4期)2022-04-13
祝您健康·文摘版(2019年3期)2019-06-11
饮食与健康·下旬刊(2017年4期)2017-05-26
中文信息(2017年2期)2017-04-13
江苏农业科学(2016年11期)2017-03-21
现代家庭·生活版(2016年11期)2016-11-09
智能计算机与应用(2016年4期)2016-09-26
古代文明(2014年1期)2014-02-23
文学教育·中旬版(2012年4期)2013-02-01
