
超高效液相色谱-串联质谱法测定叶下珠中7种成分的含量
2021-09-01 03:13秦鹏柏玉冰唐锋李洪权湖南中医药大学第一附属医院药学部长沙40007株洲市食品药品检验所湖南株洲42000
中南药学 2021年7期
秦鹏,柏玉冰,唐锋,李洪权(.湖南中医药大学第一附属医院药学部,长沙 40007;2.株洲市食品药品检验所,湖南 株洲 42000)
叶下珠为大戟科叶下珠属植物叶下珠Phyllanthus urinariaL.的干燥全草,收载于各地方标准,性微苦、凉,归肝、脾经,具有清热、利湿、解毒、消肿的功效[1-4]。叶下珠含有木脂素[5]、香豆素[6]、萜类[7-8]、黄酮[9]、酚酸[10]、有机酸[11]、生物碱[12]等成分,具有抗病毒[13]、抗肿瘤[14]、抗菌[15]、抗氧化[16]及护肝[17]等药理作用。
根据已有活性研究可知,抗病毒的作用成分为柯里拉京等[18],抗菌的作用成分为柯里拉京、没食子酸和原儿茶酸等[19-20],抗肿瘤的作用成分为柯里拉京、芦丁、槲皮素和山柰酚等[21-23],抗氧化的作用成分为柯里拉京、没食子酸、原儿茶酸、槲皮素和山柰酚等[24-26],护肝的作用成分为短叶苏木酚等[27]。
为准确把握叶下珠的用法用量,对这些功效成分进行含量测定是必不可少的。目前已有研究中对这些成分建立了含量测定方法[28-33],但这些方法仍有可改进之处,如在分析多成分时为达到一定分离度,信号采集时间较长[28-29];单次分析成分数量较少[30-32];方法灵敏度不高[33]等。这些都可能影响含量测定的高效性和准确性。而基于质荷比分析的色谱-串联质谱法几乎不依赖于峰与峰之间的分离度,信号采集时间短,灵敏度高[34],可优化上述问题。且目前尚未发现有基于此方法对叶下珠中上述功效成分进行含量测定的相关报道。
因此,本研究特基于超高效液相色谱-串联质谱(UPLC-MS/MS)法建立叶下珠中7 种功效成分的含量测定方法,以期更高效、准确地测定其含量。
1 材料
1.1 试药
槲皮素(纯度:97.4%,批号:100081-200907)、山柰酚(纯度:95.9%,批号:110861-200808)、芦丁(纯度:90.5%,批号:100080-200707)、原儿茶酸(纯度:99.9%,批号:110809-201205)、没食子酸(纯度:91.5%,批号:110831-201906)(中国食品药品检定研究院);柯里拉京(北京坛墨质检科技有限公司,纯度:98.0%,批号:A0647);短叶苏木酚(长沙诚邦生物科技有限公司,纯度:98.0%,批号:20032403);乙腈、甲醇(色谱纯,霍尼韦尔有限公司);甲酸、乙酸铵(分析纯,国药集团化学试剂有限公司)。叶下珠样品信息见表1。所有批次药材经株洲市食品药品检验所胡冠宇主管药师鉴定均为大戟科叶下珠属植物叶下珠Phyllanthus urinariaL.的干燥全草。所有批次药材均经粉碎后过4 号筛,备用。

表1 样品信息Tab 1 Information of sample
1.2 仪器
XSE 205DV 电子天平(精度:0.01 mg,梅特勒托利多仪器有限公司);HNS-SY-150 超声波仪(40 kHz,400 W,北京恒奥德仪器仪表有限公司);Agilent 1290 Infinity Ⅱ超高效液相色谱仪(安捷伦科技有限公司);QTRAP 4500 三重四极杆质谱(上海爱博才思分析仪器有限公司)。
2 方法与结果
2.1 色谱条件
采用 Welch xtimate C18色谱柱(150 mm×4.6 mm,5 μm),以含10 mmol·L-1乙酸铵的0.1%甲酸水溶液(A)-乙腈(B)为流动相,梯度洗脱(0~6 min,10%→85%B;6~8 min,85%→95%B;8~8.5 min,95%→10%B;8.5~10.0 min,10%B),流速0.8 mL·min-1,采集时间为10 min,柱温为30℃,进样量2 μL。
2.2 质谱条件
电喷雾电离源(ESI),负离子扫描,多反应监测(MRM),离子源温度:550 ℃,电喷雾电压:-4500 V,气帘气:35.0 psi,离子源气1:55 psi,离子源气2:55 psi,碰撞气:氩气。各成分的质谱参数见表2。
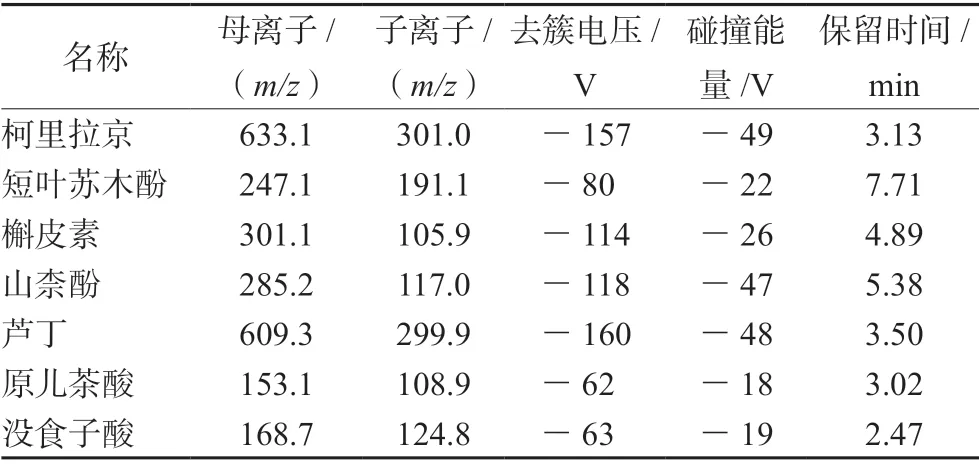
表2 负离子模式下7 种成分的保留时间及质谱参数Tab 2 Retention time and UPLC-MS/MS parameters of the 7 components in the neagtive ion mode
2.3 溶液的制备
2.3.1 混合对照品储备液 精密称取不同质量的柯里拉京、短叶苏木酚、槲皮素、山柰酚、芦丁、原儿茶酸、没食子酸对照品,置25 mL 棕色量瓶中,加70%甲醇溶解并定容至刻度,摇匀,即得。经含量折算,上述各对照品的质量浓度分别为0.500、0.260、0.130、0.500、0.320、0.700、0.300 mg·mL-1。对照品总离子流和提取离子色谱图见图1。
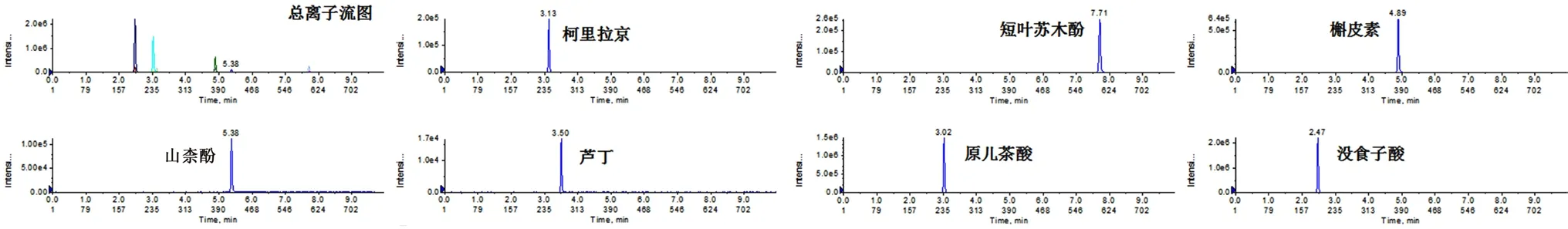
图1 叶下珠混合对照品总离子流和提取离子色谱图Fig 1 Total ion current chromatogram and extracted ion chromatogram of Phyllanthus urinaria L.mixture reference
2.3.2 供试品溶液 取叶下珠粉末(粉碎后过4 号筛),置具塞锥形瓶中,精密加入70%甲醇溶液40 mL,称量,超声(400 W,40 kHz)提取20 min,静置至室温,再称量,用70%甲醇溶液补足减失的量,摇匀,滤过,取续滤液作为供试品溶液。叶下珠样品总离子流和提取离子色谱图见图2。
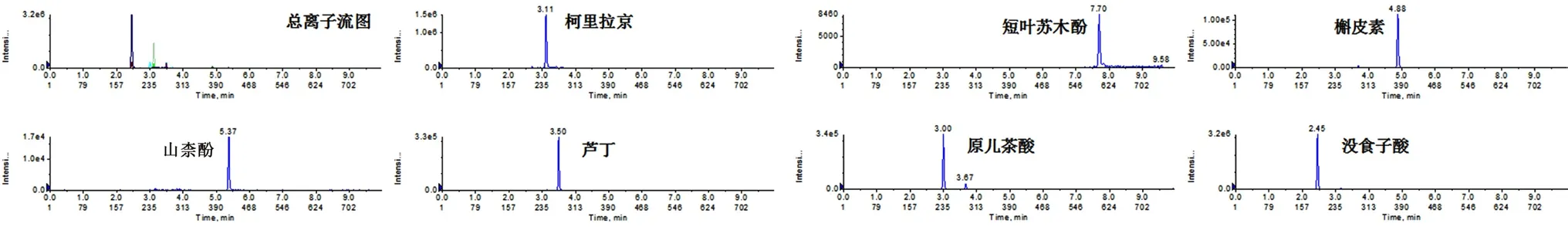
图2 叶下珠样品总离子流和提取离子色谱图Fig 2 Total ion current chromatogram and extracted ion chromatogram of Phyllanthus urinaria L.sample
2.4 方法学考察
2.4.1 线性关系考察 精密量取“2.3.1”项下混合对照品储备液适量,用70%甲醇溶液稀释得系列质量浓度的混合对照品溶液(柯里拉京:0.500~50.0 μg·mL-1;短叶苏木酚:0.0260~2.60 μg·mL-1;槲皮素:0.001 30~0.130 μg·mL-1;山柰酚:0.005 00~0.500 μg·mL-1;芦丁:0.320~32.0 μg·mL-1;原儿茶酸:0.007 00~0.700 μg·mL-1;没食子酸:0.300~30.0 μg·mL-1),按“2.1”和“2.2”项下条件进行测定,以质量浓度C(ng·mL-1)为横坐标,峰面积A为纵坐标,计算7 个成分的线性回归方程,结果见表3。

表3 线性关系结果(n=7)Tab 3 Linearity of the 7 components (n=7)
2.4.2 精密度试验 取混合对照品溶液,按照“2.1”和“2.2”项下条件分别连续进样6 次,记录各成分峰面积并计算RSD。结果柯里拉京、短叶苏木酚、槲皮素、山柰酚、芦丁、原儿茶酸和没食子酸峰面积的RSD分别为0.65%、0.88%、0.72%、0.89%、0.76%、0.60%和0.73%,表明仪器精密度良好。
2.4.3 重复性试验 取同一批样品(1 号样品),称取6 份,按照“2.3.2”项下方法制备供试品溶液,进样测定,记录各成分峰面积并计算RSD。结果柯里拉京、短叶苏木酚、槲皮素、山柰酚、芦丁、原儿茶酸和没食子酸峰面积的RSD分别为1.0%、1.2%、1.3%、1.5%、1.2%、1.4%和1.4%,表明方法重复性良好。
2.4.4 稳定性试验 取同一批样品(1 号样品)的供试品溶液,分别于0、2、4、8 和12 h 进样分析,记录各成分峰面积并计算RSD。结果各成分峰面积的RSD分别为2.0%、2.0%、2.0%、1.8%、2.5%、2.1%和1.9%,表明供试品溶液在12 h 内稳定。
2.4.5 加样回收试验 取已知含量的样品(1 号样品)6 份,每份约0.5 g,精密称定,分别加入绝对含量接近0.5 g 药材所含含量的对照品溶液后,按“2.3.2”项下方法制备供试品溶液,进样分析,计算加样回收率,结果柯里拉京、短叶苏木酚、槲皮素、山柰酚、芦丁、原儿茶酸和没食子酸的平均回收率分别为98.6%、103.8%、100.4%、101.3%、100.8%、99.6%和98.1%,RSD分别为1.6%、1.3%、1.7%、2.2%、1.8%、2.0%和1.6%,表明本方法回收率良好。
2.5 样品测定
精密称取6 批次不同批号的叶下珠各2 份,分别按照“2.3.2”项下方法制备供试品溶液,按照“2.1”和“2.2”项下条件进样测定,代入线性回归方程计算7 种成分含量,结果见表4。
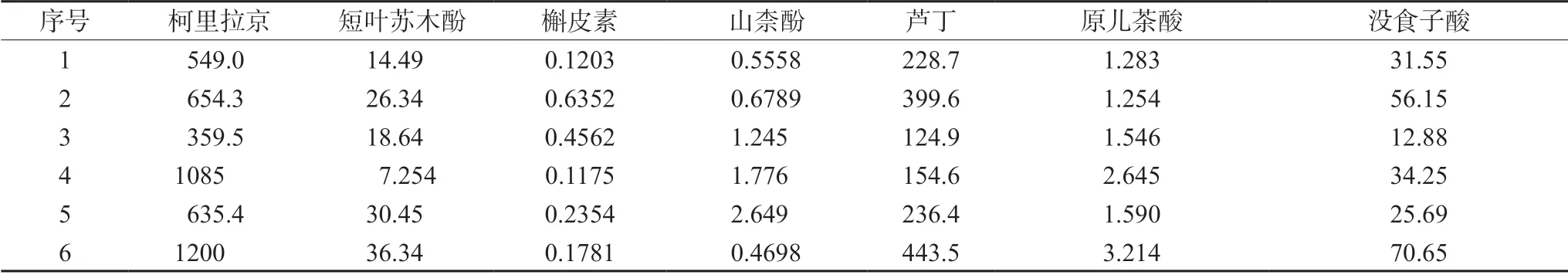
表4 6 批次样品7 种成分的含量(μg·g-1,n=2)Tab 4 Contents of the 7 components in 6 batches of samples (μg·g-1,n=2)
3 讨论
3.1 供试品溶液制备条件考察
3.1.1 提取溶剂的比较 本研究比较了30%甲醇、50%甲醇、70%甲醇、90%甲醇作为提取溶剂,以7 种成分的峰面积大小为判断依据,确定最佳提取溶剂。结果当甲醇浓度为70%时,7 种成分的峰面积均达到最大值。因此,选择70%甲醇为提取溶剂。
3.1.2 提取时间的比较 本研究比较了10、20和30 min 的提取时间,以7 种成分的峰面积不再增加为判断依据,确定最佳提取时间。结果7 种成分均在提取20 min 时达到最大值。而当提取时间为30 min 时,部分成分含量不增反降,尤其是原儿茶酸和没食子酸,这可能是由于随着超声时间的增加,水温升高,促使部分酚酸被氧化。因此,20 min 为最佳提取时间。
3.1.3 提取体积的比较 本研究比较了30、40和50 mL 的提取体积,以7 种成分的峰面积与其提取所加入溶剂体积的乘积达到上限为判断依据,确定最佳提取体积。结果发现当提取体积为40 mL 时,其峰面积与提取时所加入溶剂的体积的乘积已经达到上限,因此,40 mL 为最佳提取体积。
3.2 色谱条件优化
3.2.1 色谱柱的选择 本研究比较了两种不同的色谱柱,Welch Xtimate C18(4.6 mm×150 mm,5 μm)和Thermo Acclaim 120 C18(4.6 mm×150 mm,5 μm),以7 种成分的分子离子峰的保留性和峰形等因素为判断依据,确定更为合适的色谱柱。根据试验结果,两款色谱柱均获得了较为合适的保留时间和较好的峰形,但仍有区别。当使用Thermo Acclaim 120 C18柱时,原儿茶酸、没食子酸、短叶苏木酚和柯里拉京峰稍有前延,槲皮素和山柰酚峰稍有拖尾;而当使用Welch Xtimate C18色谱柱,峰形均未出现峰拖尾和前延的现象。因此,选择Welch Xtimate C18色谱柱较为合适。
3.2.2 流动相系统的选择 本研究比较了3 种不同的流动相系统,包括乙腈-水、乙腈-0.1%甲酸水溶液和乙腈-含10 mmol·L-1乙酸铵的0.1%甲酸水溶液,以7 种成分的分子离子峰的峰形为判断依据,确定更为合适的流动相系统。结果发现当选择乙腈-水作为流动相时,槲皮素、没食子酸稍有拖尾;当选择乙腈-0.1%甲酸溶液水作为流动相时,短叶苏木酚稍有拖尾;而选择以乙腈-含10 mmol·L-1乙酸铵的0.1%甲酸水溶液作为流动相时,峰形均较为对称。因此,选择乙腈-含10 mmol·L-1乙酸铵的0.1%甲酸水溶液较为合适。
3.2.3 水相起始比例的选择 本研究比较了水相90%、70%和50% 3 种不同起始比例,以7 种成分的分子离子峰的峰形为判断依据,确定最佳水相起始比例。发现不同起始比例情况下,峰形差异较大。当水相起始比例为90%时,各峰峰形较好;当起始比例降低至70%时,7 种成分的峰形变差,出现不同程度的峰前延和拖尾现象;当降低至50%时,峰前延和拖尾现象加剧,尤其是原儿茶酸、没食子酸的峰前延和槲皮素、山柰酚峰拖尾现象,甚至,柯里拉京出现峰分叉的现象。因此,选择90%的水相作为起始比例较为合适。
3.3 总结
综上,基于上述供试液制备条件的考察、色谱条件的优化、质谱参数的建立及方法学考察等,本研究首次建立了叶下珠中柯里拉京、短叶苏木酚、槲皮素、山柰酚、芦丁、原儿茶酸和没食子酸7 种成分的UPLC-MS/MS 含量测定方法。本方法的完成,可更为高效、准确地测定叶下珠中多种成分的含量,为基于这些成分的叶下珠药用价值开发奠定了研究基础。
猜你喜欢
现代食品科技(2022年5期)2022-05-30
中成药(2021年7期)2021-09-27
疯狂英语·新读写(2018年3期)2018-11-29
天然产物研究与开发(2018年8期)2018-09-10
中成药(2017年9期)2017-12-19
中成药(2017年3期)2017-05-17
中国病理生理杂志(2015年8期)2015-12-21
读者·校园版(2014年21期)2014-05-14
食品科学(2013年13期)2013-03-11
长江大学学报(自科版)(2013年33期)2013-03-11
