
薏苡转录组测序及基因功能注释
2021-09-01 03:13:44欧阳雨晴李玲玲石好宇赵艺朱甫臻尹艳燕王灵芝北京中医药大学生命科学学院北京100029
中南药学 2021年7期
欧阳雨晴,李玲玲,石好宇,赵艺,朱甫臻,尹艳燕,王灵芝(北京中医药大学生命科学学院,北京100029)
转录组(transcriptome)是指细胞在特定发育阶段或生理条件下,所表达的全部RNA 的总和,可反映相同基因在不同环境下表达的差异,揭示不同基因间的相互作用及其各自的功能[1-2]。转录组测序,又称为RNA 测序(RNA-seq),将样本总RNA 反转录成cDNA 后,进行高通量测序来确定样本中整体转录组的表达情况。自2005年以来,以Solexa 技术、SOLiD 技术和454 技术为代表的第二代测序技术,推动了RNA-seq 技术的迅速发展。RNA-seq 可在限定时间内全面而快速地获取特定样品的几乎所有mRNA 序列信息,该技术在基因功能分析、特异表达基因挖掘、信号途径构建、代谢途径鉴定、基因家族确定等方面发挥重要作用[3]。RNA-seq 技术已经成功应用于许多药用植物转录组信息的收集,如黄连[4]、人参[5]、黄三七[6]、银杏[7]、红豆杉[8]和阿育魏实[9]等,在植物有效成分生物合成的功能基因挖掘和分子标记育种方面取得了较大进展[10]。
薏苡(Coix lacryma-jobiL.var.mayuenStapf),是禾本科(Gramineae)玉蜀黍族(Maydeae),薏苡属CoixL.植物[11],为药食同源作物,具有利水渗湿、健脾止泻之功能,中医临床常用于治疗水肿、排尿困难和腹泻等[12]。薏苡仁含有多种化学成分如多糖、蛋白质、脂质、多酚、类胡萝卜素、植物甾醇、螺甾内酯和内酰胺[13],具有多种药理功能,如抗肿瘤[14]、免疫调节[15]、抗氧化[16]、降血压[17]等功能。其中,薏苡仁油因具有显著的抗肿瘤活性,已被研制成康莱特注射液,广泛应用于临床,并被美国食品和药物管理局(FDA)批准在美国进行临床试验[18]。薏苡仁油的主要活性成分为三酰甘油类,占薏苡仁油含量的86.96%~94.39%[19],其中甘油三油酸酯为《中国药典》(2020年版)薏苡仁质量标准的指标性成分。研究表明1,3-二油酸-2-亚油酸甘油酯[20]和1-棕榈酸-2-亚油酸-3-油酸甘油酯[21]具有显著抗肿瘤活性。根据溶解度不同,种子储藏蛋白分为清蛋白、球蛋白、醇溶蛋白和谷蛋白,谷蛋白约占总蛋白含量的40%[22],具有多种潜在的生物活性。美国山核桃谷蛋白源多肽有降压,抗氧化和抗肿瘤活性[23]。苋菜谷蛋白水解产物有降血糖作用[24]。胡桃楸谷蛋白水解产物具有免疫调节作用[25]。本课题组前期研究表明,薏苡仁谷蛋白源小分子肽具有免疫调节活性[26]。
薏苡仁的生物活性主要取决于其品种。薏苡在中国除青海和宁夏以外的大多数省份中均有分布,药材的质量与生产区域的环境密切相关[27],由于地理环境和栽培条件的差异,使得我国薏苡仁种质资源极为丰富多样。但薏苡基因序列信息缺乏,阻碍其遗传育种与药用价值的开发。本研究采用Illumina Hiseq 测序平台技术,构建薏苡转录组数据库,并进行功能注释,为谷蛋白和三酰甘油等成分合成关键基因提供序列信息。
1 材料与方法
1.1 药材
薏苡种植于北京中医药大学的药材种植园,采集不同发育时期的根、叶、穗、幼苗及籽粒,液氮冷冻后,-80℃保存,用于后续RNA 提取。
1.2 仪器与试药
EASY spin 植物RNA 快速提取试剂盒(北京博迈德生物技术有限公司);1%琼脂糖凝胶电泳检验RNA 完整性;Qubit Fluorometer 荧光定量仪(美国BioTek);NanoDrop 超微量分光光度计(美国Thermo);Agilent 2100 生物分析仪(美国安捷伦科技有限公司)。
1.3 转录组文库的建立和测序分析
样品检测合格后,等量混合EASY spin 植物RNA 快速提取试剂盒提取的各薏苡组织的总RNA,经带有Oligo(dT)的磁珠纯化mRNA 以构建cDNA 文库。 采用Illumina HiSeq 4000 以PE 100 的测序读长对cDNA 进行测序,得到原始数据(Raw reads)。使用SOAPnuke 处理Raw reads,去除低质量,包含接头以及未知碱基N含量大于5%的reads,得到Clean reads。使用Trinity 对Clean reads 进行De novo组装,使用Tgicl 对组装的转录本进行聚类,去除冗余获得unigene。N50 值用于评估组装过程中的连续性。
1.4 转录本功能注释
将组装得到的薏苡转录本数据,通过BLAST 软件分别与NT、NR、COG、KEGG 以及SwissProt 数据库进行比对;基于NR 注释,使用Blast2GO 软件获得GO 注释,使用InterProScan5软件获得InterPro 注释。
1.5 生物信息学分析
使用Blast 软件将unigene 序列按照 NR、SwissProt、KEGG、COG 数据库的优先顺序进行比对,得到unigene 编码区5'-3'方向的核酸序列,即CDS。使用ESTScan 软件对未比对上的unigene 序列,进行CDS 预测。使用MISA 软件对unigene 进行SSR 分析,并用Primer 3.0 软件设计SSR 引物。使用HISAT 软件将Clean reads 比对到unigene,然后通过变异检测软件GATK 识别SNP 位点。
1.6 基因表达量计算
基因表达量用 FPKM(Fragments Per Kilobase of transcript per Million mapped reads)表示。使用Bowtie2 软件将Clean reads 比对到unigene,然后利用RSEM 计算各样品的基因表达水平。FPKM计算公式如下:
FPKM=cDNA Fragments/[Mapped Fragments(Millions)×Transcript Length(kd)]
式中,cDNA Fragments 表示比对到某一转录本上片段数目,即双端Reads 数目;Mapped Fragments(Millions)表示比对到转录本上的片段总数,以106为单位;Transcript Length(kd)表示转录本的长度,以1×103个碱基为单位。
2 结果
2.1 测序结果与序列组装
电泳图谱(见图1)表明样品RNA 完整性较好。根据HiSeq 测序样品质量标准,各样品均为A级,满足后续cDNA 文库构建要求。使用Illumina HiSeq 4000 平台共得到了65.60 Mb Raw reads,过滤后得到99.95%的Clean reads(65.57 Mb),Q20为96.09%。读本中碱基分布均匀没有明显的AT或GC 分离现象(见图2A),表明该RNA-seq 质量较高。使用Trinity 软件对Clean reads 进行De novo组装,共获得102 092 个transcripts。经Tgicl软件将transcrips 聚类并除去冗余之后,获得了76 164 个unigene,平均长度为1023 bp,GC 含量为49.36%,N50 为1700 bp,表明组装效果较好(见表1),其中序列长度为300 bp 数目最多,有18 321 条(见图2B)。

表1 组装过程质量指标Tab 1 Quality index of assembly process

图1 薏苡各组织RNA 电泳图Fig 1 RNA electrophoretogram of Coix tissues

图2 Clean reads 碱基含量(A)和unigene 长度(B)分布图Fig 2 Clean reads base content(A)and unigene length(B)distribution
2.2 基因功能注释
将unigene 序列与NT、NR、COG、KEGG、SwissProt、GO 和InterPro 七大功能数据库进行比对,共有59 460 个unigene 获得注释,占总数的78.07%。其中,在NT 数据库中,获得注释的unigene 数量最多,有56 121 个,占73.68%。而在COG 数据库中,获得注释的unigene 数量最少,仅有20 670 个,占27.14%(见表2)。unigene 功能注释Venn 图(见图3A)显示,共有14 177 个unigene在NR、Interpro、Swissprot、COG 和KEGG 5 个数据库同时得到注释,占unigene 总数的23.84%。

表2 Unigenes 功能注释Tab 2 Function annotation of unigenes
2.2.1 Unigene 的NR 数据库比对分析 将unigene与NR 数据库进行相似序列匹配,结果如图3B 所示,共有49 092 个unigene 在NR 数据库中注释成功。其中薏苡与高粱(Sorghum bicolor)同源的序列最多,占NR 总注释的51.73%,其次匹配的物种有玉米(Zea mays,26.29%)、小米(Setaria italica,10.16%)和水稻(Oryza sativaJaponica Group,3.21%)。
2.2.2 Unigene 的GO 功能分类分析 如图3C 所示,共有27 407 个unigene 在GO 数据库中注释成功,得到156 122 条对应关系。三大功能中生物学过程最多,为67 477 条(43.22%);细胞组分数目为55 713 条(35.69%);分子功能最少为32 932条(21.09%)。这三大功能可详细划分为56个分支,生物学过程22个、细胞组分17个、分子功能17个。在生物学过程类型中,与新陈代谢过程相关的unigene 为15 060 条,所占比例最高;在细胞组分功能分类中,细胞与细胞成分所涉及的unigene 数量一致且最多,有14 043 条;在分子功能类型中,注释上的unigene 涉及结合、抗氧化、催化、翻译调节及营养储存等多方面功能活性,其中,涉及到结合功能的unigene 有14 728 条,所占比例最高,对应蛋白质标签功能的unigene 最少(1 条)。
2.2.3 Unigene 的COG 注释结果分析 如图3D所示,共有20 670 个unigene 在COG 数据库中注释成功,根据其功能的不同可分为25 类。一般功能基因类别中unigene 数量最多,占unigene总量的27.33%;核结构类unigene 有4 条,所占比例最小。此外,翻译、核糖体结构与生物合成(19.86%),转录(17.50%),信号转导机制(12.13%),复制、重组与修复(16.72%),翻译后修饰、蛋白质转运及分子伴侣(12.76%),细胞膜、细胞壁生物合成(10.42%),细胞周期调控、细胞分裂与染色体重排(14.61%),碳水化合物转运与代谢(11.55%),氨基酸转运与代谢(7.51%)等功能类别的unigene 表达丰度也相对较高。
2.2.4 Unigene 的KEGG 代谢途径分析共有30 941 个unigene 在KEGG 数据库中注释成功,涉及126 个代谢途径(见图3E)。与新陈代谢相关的unigene 数量最多,参与影响氨基酸代谢(1363 个)、次级代谢产物代谢(1085 个)、碳水化合物代谢(2418 个)、能量代谢(704 个)、脂质代谢(3308 个)及核酸代谢(1020 个)等多个方面,其中,参与聚糖合成与代谢的基因最少,仅有496 个,占unigene 总量的1.60%。
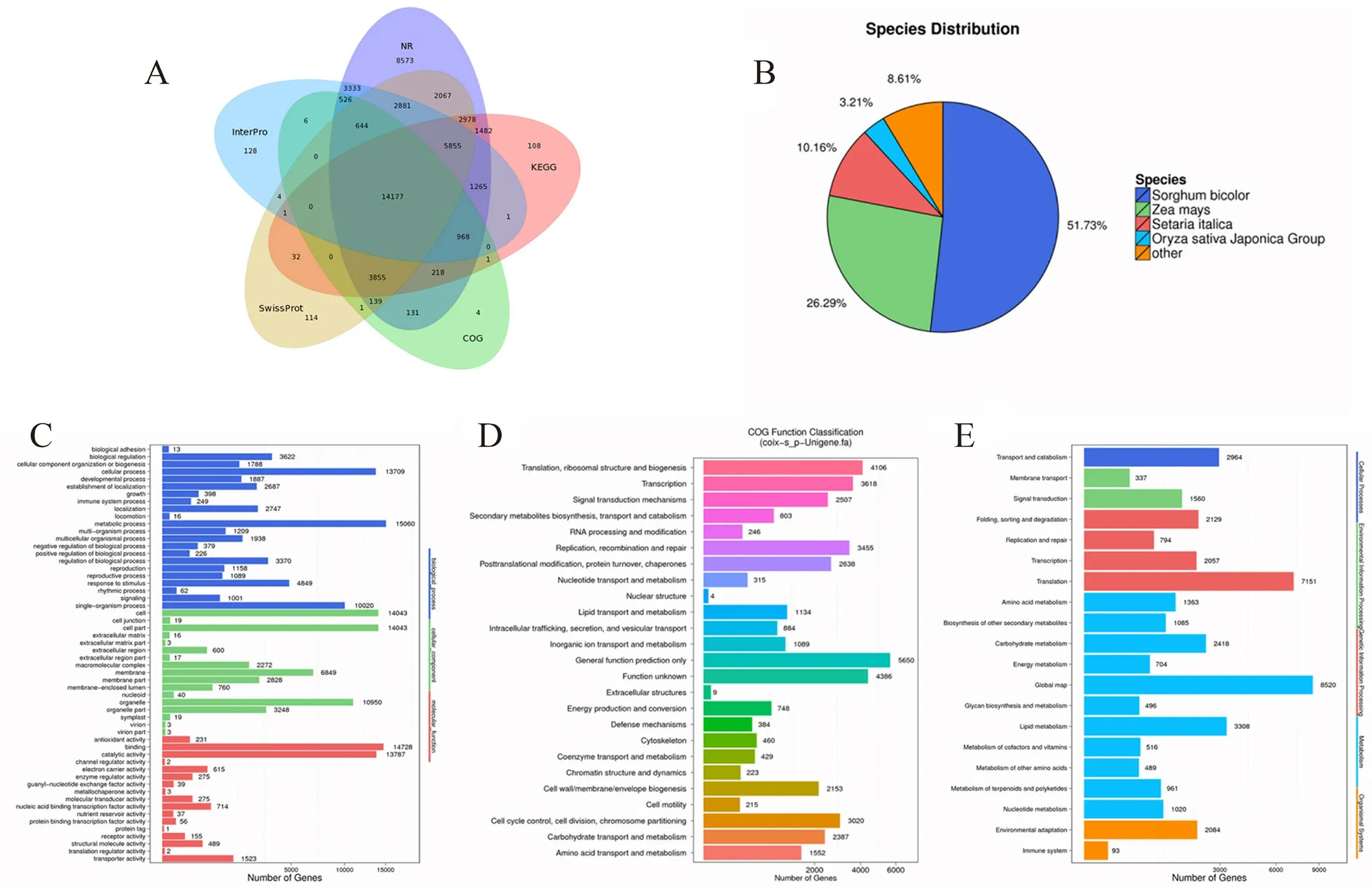
图3 Unigene 功能注释图Fig 3 Functional annotation of unigenes
经统计分析,注释到三酰甘油代谢路径(ko00561)的unigene 有388 个,结合三酰甘油目前已知的生物合成途径,筛选出257 个与三酰甘油生物合成途径相关的unigene,并对其表达量进行分析(见表3)。二酰基甘油酰基转移酶(diacylglycerolO-acyltransferase,DGAT) 是依赖酰基辅酶A 的三酰甘油从头合成途径的关键酶和限速酶。分别有19、5、1 个unigene 编码DGAT1、DGAT2、DGAT3,其中编码DGAT3 的unigene 表达量最高,FPKM 值为30.20。磷脂:二酰基甘油酰基转移酶(phospholipid:diacylglycerol acyltransferase,PDAT)是不依赖酰基辅酶A 的合成途径的关键酶,共有10 个unigene 编码PDAT,FPKM 值最高为35.49。除此之外与三酰甘油生物合成的途径相关的酶中表达量较高的还有醛脱氢酶(NAD+)[aldehyde dehydrogenase(NAD+),ALDH]、1-酰基-sn-甘油-3-磷酸酰基转移酶(1-acyl-sn-glycerol-3-phosphate acyltransferase,plsC)和酰基甘油脂肪酶(acylglycerol lipase,MGLL),其FPKM 分别为993.54、113.86、70.14。
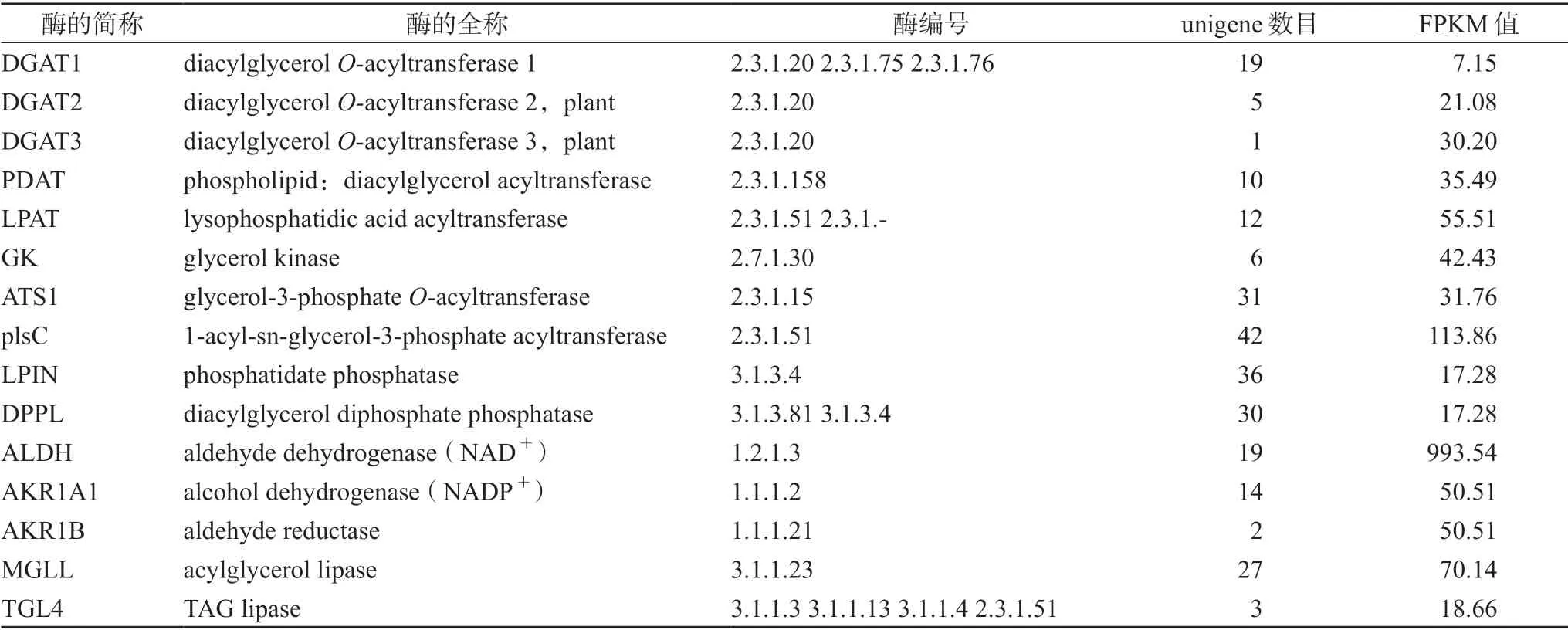
表3 薏苡三酰甘油生物合成中编码关键酶的unigene Tab 3 Unigenes encoding the key enzymes involved in the synthesis of triacylglycerol in Coix
2.3 CDS 预测
CDS 预测可为薏苡基因组图谱绘制及基因功能研究提供关键依据。使用BLAST 软件比对得到48 449 个CDS 序列(63.61%),通过ESTScan 软件预测得到2711 个CDS 序列(3.56%)。总共获得51 160 个CDS 序列,占unigene 总数的67.17%。CDS 长度分布如图4A 所示,长度在200~800 bp 内的CDS 序列有33 550 个(69.25%);长度为300 bp 的数量最多,9793 个(20.21%);其中与谷蛋白相关的CDS 序列有9 个(见表4),分别来自Swissprot、NR 数据库和NT 数据库,可用于后续谷蛋白基因克隆和定点突变研究。
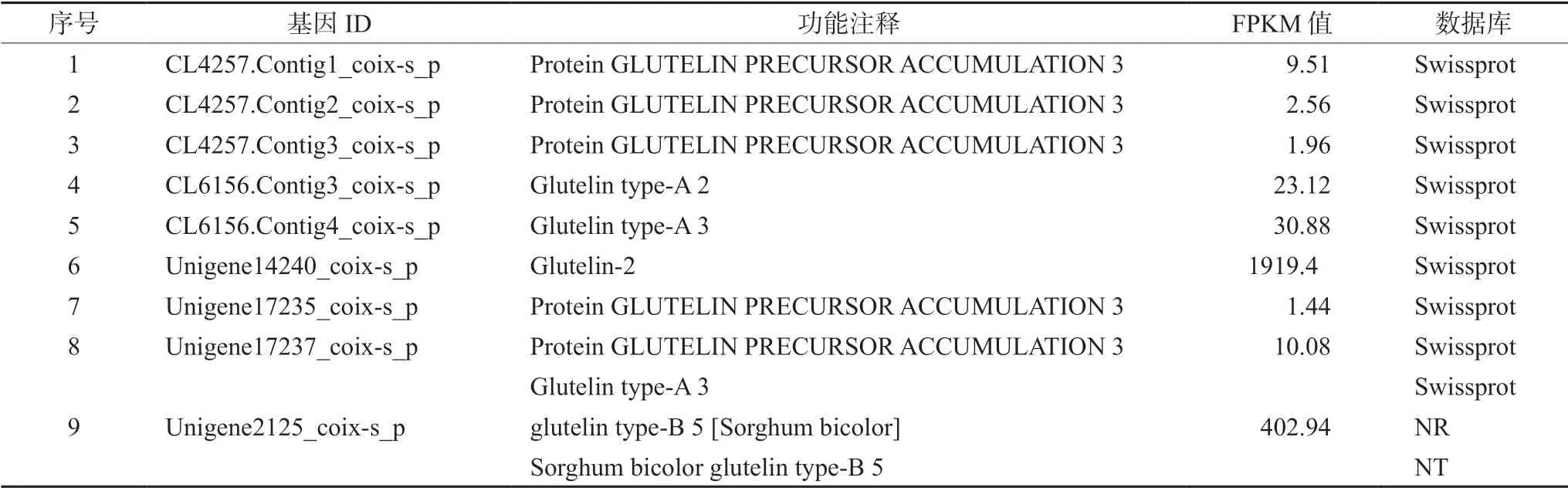
表4 薏苡谷蛋白相关CDS 序列Tab 4 Glutelin related CDS of Coix
2.4 SSR 检测结果
使用MISA 软件对unigene 进行SSR 检测,共搜索得到分布于11 101 个unigene 中的3324 个SSR,占unigene 总序列的29.94%。SSR 长度特征见表5及图4B,SSR 类型丰富,从单核苷酸到六核苷酸均有出现,其中,三核苷酸重复最多(7899,占71.2%),出现频率最高的基序为CCG/CGG(3690),最低的是ACT/AGT(84)。在二核苷酸重复类型中,AG/CT 基序出现的频率最高(1407),而CG/CG 出现的频率最低(242)。

图4 薏苡CDS(A)和SSR(B)分布图Fig 4 CDS(A)and SSR(B)distribution in Coix

表5 SSR 长度统计结果Tab 5 Statistics of SSR length distribution
2.5 SNP 位点分析结果
使用GATK 软件对样本基因进行SNP 检测,共鉴定出22 764 个SNP,其中转换15 141个(66.51%),发生A-G 替换的有7704 个,发生C-T 替换的有7437 个;变异类型为颠换的共7623 个(33.49%),发生A-C、A-T、C-G 及G-T替换的数目分别为1831、1699、2322 及1771 个(见表6)。
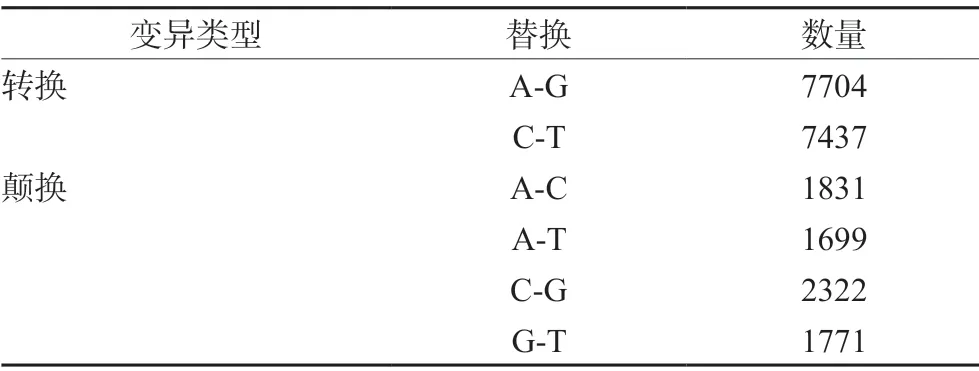
表6 SNP 变异类型统计Tab 6 Statistics of SNP variation types
2.6 Unigene 表达量计算
根据组装结果,使用 Bowtie2 软件将Clean reads 比对到unigene,后使用RSEM 软件计算样品的基因表达水平,计算获得72 188 条unigene的表达量。
3 讨论
RNA-seq 技术具有灵敏度高、通量高、成本低、操作简单、重复性好、检测范围广、分辨率高等诸多优势,不局限于已知的基因组序列信息,还可用于发现未知基因及非编码RNAs,精确地识别可变剪切位点以及SNP,提供更加全面的样本转录组信息[3,28-29]。目前,RNA-seq 技术已成功应用于多种药用植物转录组信息的采集。李依民等[6]应用Illumina HiSeq 2000 测序平台,以PE150 的测序读长,对黄三七根茎进行RNA-seq 技术,得到63 322 086 条Clean reads 和52 575 个unigene,为今后黄三七功能基因的鉴定、代谢途径的分析及奠定基础。Majeed 等[8]构建了红豆杉参考转录组,经注释得到129 869 个unigene,同时产生了7041 个SSR 标记,为破译红豆杉遗传多样性及其保护提供了有价值的信息。Amiripour 等[9]对阿育魏实的花序进行RNA-seq 技术,De novo组装后产生68 051 个unigene,其中43 156 个获得注释,生成了8751 个SSR 标记,是目前第一个可用于转录组表征、基因发现、开发SSR 分子标记和阿育魏实基因组研究的数据库。邓楠等[30]应用Illumina HiSeq 2000 测序平台对膜果麻黄不同发育期种子进行RNA-seq 技术,得到12 999 122 条初始序列,组装后获得49 449 条编码基因,并根据KEGG 通路找到了有关姜醇烷、二芳基庚及芪类合成途径的编码基因片段230 个。张绍鹏等[31]获得了珠子参根茎的转录组信息数据,为该物种资源的有效开发和利用提供了重要的理论基础。李铁柱等[32]利用高通量测序技术构建杜仲叶片和果实的转录组数据库,共获得54 471 338 条Raw reads,拼接组装后得到49 610 条unigene,总长度为37 616 729 bp。本研究使用Illumina HiSeq 4000平台对薏苡不同发育时期的各种组织部位RNA进行RNA-seq 技术,构建薏苡转录组数据库。总共获得76 164 个unigene,总长度为77 917 702 bp,数据量大且组装效果较好,可为薏苡分子生物学研究及相关药用价值的开发提供重要依据。将unigene 与七大功能数据库比对后分别有49 092(NR:64.46%)、56 121(NT:73.68%)、32 748(Swissprot:43.00%)、20 670(COG:27.14%)、30 941(KEGG:40.62%),27 407(GO:35.98%)以及 29 789(Interpro:39.11%)个unigene 得到注释。未得到注释的unigene 占21.93%,推测原因可能是由于数据库基因信息缺乏、测序技术的局限、序列长度较短未包含蛋白质功能域,或者是由于序列本身是非编码序列或不完整序列或薏苡特有的新基因[33]。NR 数据库比对结果显示,薏苡与高粱具有最近的亲缘关系(51.73%),这与Ottoboni等[34]研究结果一致。GO 功能分类中涉及代谢过程的unigene 数量最多(15 060)与KEGG 数据库的注释结果相一致。由KEGG 注释结果可知,代谢分支中,除涉及全局和概览图途径外,涉及脂质代谢途径的unigene 数量最多(3308),提示脂类化合物在薏苡生长发育过程中有着重要作用。本研究分析了三酰甘油生物合成的相关代谢通路的257 个unigene,为今后提高三酰甘油含量奠定了理论基础。
质谱鉴定中的数据库搜索是肽/蛋白质结构解析的重要步骤,通过扩展蛋白数据库可显著提高肽/蛋白质鉴定的准确性和灵敏度。Goecks 等[35]采用RNA-seq 技术建立了果蝇雌性寄生蜂腹部转录本序列文库,液相色谱-串联质谱法鉴定寄生蜂毒液腺腔分离纯化的肽序列,将肽谱数据与转录组数据库信息进行比对,最终从寄生蜂L.boulardi毒液中鉴定获得了129 种蛋白,从L.heterotoma毒液中鉴定得到176 种蛋白。本研究共获得51 160 个CDS 序列,成功构建了薏苡蛋白数据库,解决了以往薏苡蛋白序列信息量较少的问题,丰富了禾本科植物蛋白数据库,为薏苡及其近缘物种蛋白及多肽序列的分析、鉴定提供重要依据。薏苡仁谷蛋白水解物具有显著的降压活性,本研究中获得的CDS 序列中有9 个与谷蛋白相关,借助分子标记辅助育种,选择高表达谷蛋白相关基因亲本材料,可为薏苡抗高血压亲本的选育,提高薏苡仁的药用价值奠定了理论基础。推测到的3324 个SSR中CCG/CGG 基序出现最多(36 090),与水稻、高粱、玉米的SSR 检测结果相一致[36]。基于这3324个SSR 位点,开发SSR 分子标记,将为薏苡的遗传多样性研究和进化研究奠定基础。得到的22 764个SNP 位点中,转换位点以A-G 居多(7704),颠换位点最多的是C-G(2322),与张得芳等[37]对花叶海棠的SNP 位点研究相一致。薏苡生长发育的过程中需要大量基因的调控,而SNP 位点的改变可能会导致所在基因序列的改变,从而对基因的转录、翻译过程造成影响,得到结构和功能可能发生变化的蛋白质,进而造成薏苡植株性状的改变[38]。因此,通过对SNP 位点和其改变所造成的植株性状变化,可为薏苡的分子育种提供理论参考。
猜你喜欢
中华胰腺病杂志(2023年5期)2023-06-05 23:35:48
广东第二课堂·小学(2023年5期)2023-05-29 02:16:13
现代农业科技(2022年14期)2022-12-14 14:16:21
中国油脂(2022年1期)2022-02-12 09:42:50
光谱学与光谱分析(2021年11期)2021-11-11 05:22:46
中国食品学报(2021年3期)2021-04-22 06:25:38
食品与生物技术学报(2021年6期)2021-01-15 22:39:59
中国粮油学报(2020年10期)2020-11-05 03:09:06
农技服务(2020年2期)2020-05-20 09:55:12
医学综述(2020年8期)2020-02-16 08:34:45
