
高效液相色谱法测定富马酸比索洛尔原料药及其制剂中的有关物质
2021-09-01 03:13程智王孝艳雍子宜谢华四川省药品检验研究院成都673成都医学院药学院成都60500
中南药学 2021年7期
程智,王孝艳,雍子宜,2,谢华*(.四川省药品检验研究院,成都 673;2.成都医学院药学院,成都60500)
富马酸比索洛尔是目前临床上治疗心血管疾病常见的一种选择性β1-肾上腺素受体拮抗剂[1-3],在《中国药典》2020年版(ChP2020)、美国药典(USP42)、欧洲药典(EP10.0)、英国药典(BP2020)以及日本药典(JP17)中均有收载。富马酸比索洛尔片在ChP2020、美国药典(USP42)、英国药典(BP2020)以及日本药典(JP17)中均有收载,国内有批准文号的3 家生产企业均通过了仿制药一致性评价研究,并获批了新的药品标准[4-7]。富马酸比索洛尔胶囊为我国特有剂型,只在ChP2020 中收载。
在有关物质检测方面,除美国药典(USP42)在片剂中无规定外,其他标准均为有关物质检测提供了方法和参考,但各标准在已知杂质、色谱条件和杂质限度等方面均存在着很大的差异,特别是在国内各企业通过仿制药一致性评价获得新的注册标准后,其有关物质测定方法及限度均高于ChP2020,故存在注册标准与药典并行检验的情况。为统一和提高我国药品标准,参考国内外各标准中有关物质的测定方法,结合国内原料药及其制剂中杂质含量的实际情况,建立HPLC 法测定富马酸比索洛尔及其制剂中的有关物质。
1 仪器与试药
1.1 仪器
Waters 2695 高效液相色谱仪(美国Waters公司);岛津LC-20AD 高效液相色谱仪(岛津中国有限公司);CPA225D 十万分之一电子天平、XPR6UD5 百万分之一电子天平、SevenExcellence 多参数测量酸度计(梅特勒-托利多公司);Sartorius arium pro 纯水仪(德国Sartorius 公司)。
1.2 试药
富马酸比索洛尔对照品(批号:100711-201602,纯度:99.7%)、富马酸对照品(批号:111541-201803,纯度:99.9%)(中国食品药品检定研究院),杂质A(批号:A-J20161128-01,纯度:99.7%)、杂质B(批号:LGC,MM0460.07,纯度:100%)、杂质E(批号:19085,纯度:99.5%)、杂质G(批号:1455-093A2,纯度:99.2%)、杂质K(批号:3015-0692A2,纯度:99.3%)、杂质L(批号:150401,纯度:97.1%)、杂质M(批号:VST019,纯度:97.0%)、杂质N-醛(批号:VST009,纯度:97.0%)、杂质酸(批号:VST020,纯度:97.0%)、杂质羟乙酯(批号:VST021,纯度:97.0%)(自制)。磷酸二氢铵(分析纯)、磷酸(分析纯)、乙腈(色谱纯)、水为纯化水。
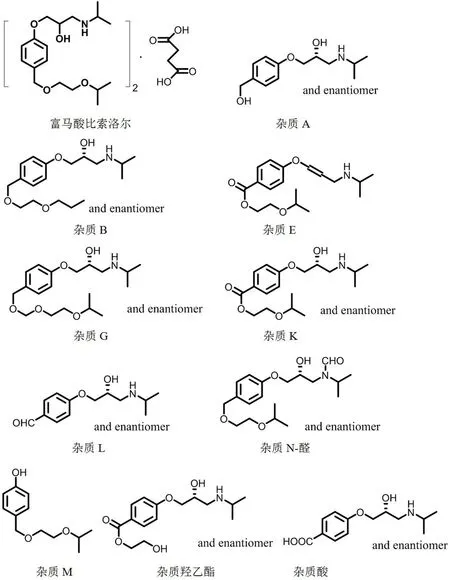
图1 富马酸比索洛尔及其杂质化学结构式Fig 1 Chemical structure formula of bisoprolol fumarate and its impurity
富马酸比索洛尔原料药(企业A,批号:1712241;企业B,批号:9351059、9351060、9351061;企业C,批号:20160202、180402、181201);富马酸比索洛尔片(企业A,批号:1903115、1808281;企业B,批号:NT001201、NT001882;企业C,批号:200306、180802);富马酸比索洛尔胶囊(企业D,批号:190101、190201、190702,规格:2.5 mg)。
2 方法与结果
2.1 色谱条件
色谱柱Kromasil C18(4.6 mm×250 mm,5 μm),流动相A 为磷酸盐缓冲液(称取磷酸二氢铵3.45 g,加水900 mL,磷酸调节pH 值至2.5,加水至1000 mL),流动相B 为磷酸盐缓冲液-乙腈(20∶80,V/V),梯度洗脱,流速1.0 mL·min-1,检测波长225 nm,柱温30℃,进样量20 μL。梯度洗脱程序见表1。
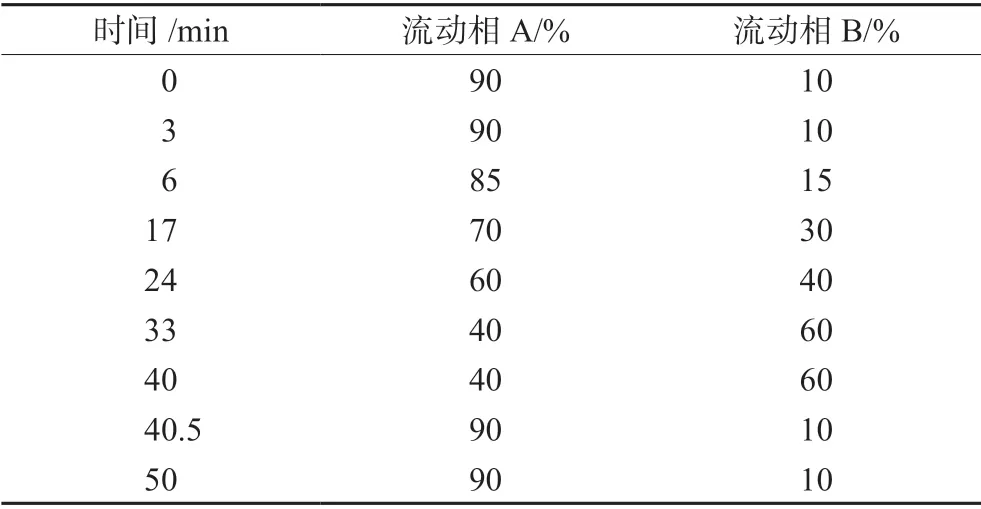
表1 梯度洗脱程序Tab 1 Gradient elution program
2.2 溶液的制备
2.2.1 对照品溶液的制备 分别称取杂质A、杂质酸、杂质L、杂质羟乙酯、杂质G、杂质K、杂质M、杂质N-醛和富马酸比索洛尔对照品适量,用溶剂(磷酸盐缓冲液)溶解并定量稀释制成每1 mL 约含比索洛尔1 μg、杂质A 1.5 μg、杂质羟乙酯和杂质酸2 μg、其他杂质2.5 μg 的混合对照品溶液,即得。
2.2.2 供试品溶液的制备 取本品细粉适量(约相当于富马酸比索洛尔25 mg),精密称定,置50 mL 量瓶中,加溶剂溶解并稀释至刻度,摇匀,滤过,取续滤液即得。
2.2.3 对照溶液的制备 精密量取“2.2.2”项下供试品溶液1 mL,分别置于100 mL 量瓶中,加溶剂稀释至刻度,摇匀,即得。
2.2.4 系统适用性试验溶液 分别称取杂质A、杂质酸、杂质L、杂质羟乙酯、杂质G、杂质K、杂质M、杂质N-醛和富马酸比索洛尔对照品适量,用溶剂溶解并定量稀释制成每1 mL 约含比索洛尔500 μg 和其他各已知杂质2 μg 的溶液,即得。
2.2.5 空白辅料溶液 取混合辅料约0.75 g,置50 mL 量瓶中,加溶剂溶解并稀释至刻度,摇匀,滤过,取续滤液即得。
2.3 方法学考察
2.3.1 系统适用性试验 精密量取“2.2.4”项下的系统适用性溶液,按“2.1”项下色谱条件进样分析,记录色谱图。结果空白辅料峰无干扰,比索洛尔峰与各杂质峰分离度均>1.5,理论板数均>3000,见图2。
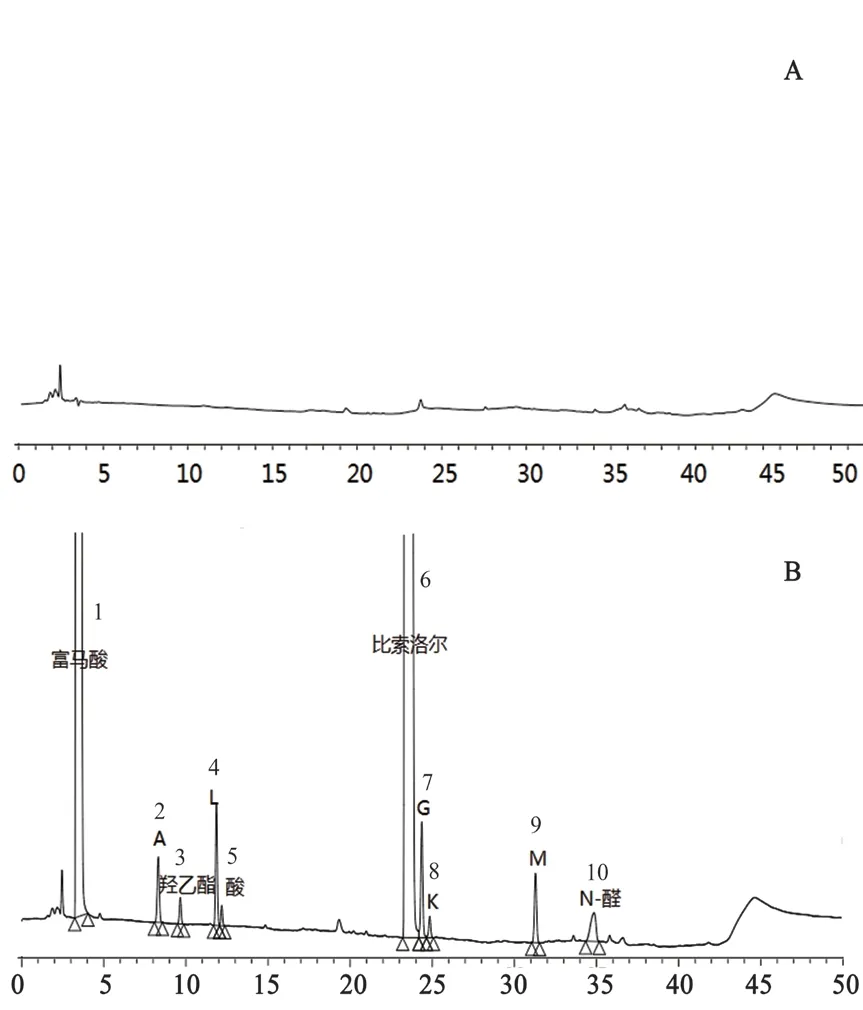
图2 系统适用性试验色谱图Fig 2 Chromatogram of system suitability
2.3.2 专属性试验 取富马酸比索洛尔原料(企业B,批号:9351060)约25 mg,加混合辅料(均为片剂与胶囊的混合辅料)0.75 g 置50 mL 量瓶中,分别按以下方式处理:
① 加水溶解稀释至刻度,摇匀,作为未破坏供试品溶液;② 酸破坏:加入1 mol·L-1盐酸溶液2 mL,室温放置1.5 h 后,加入1 mol·L-1氢氧化钠溶液2 mL 中和,加水稀释至刻度,摇匀,作为酸破坏供试品溶液;③ 碱破坏:加2 mol·L-1氢氧化钠溶液2 mL,100℃水浴加热3 h,加入2 mol·L-1盐酸溶液2 mL 中和,加水稀释至刻度,摇匀,作为碱破坏供试品溶液;④ 氧化破坏:加30%的过氧化氢溶液2 mL,水浴80℃加热2 h,加水稀释至刻度,摇匀,作为氧化破坏供试品溶液;⑤ 高温破坏:加水适量溶解,于100℃水浴加热4 h,加水稀释至刻度,摇匀,作为高温破坏供试品溶液;⑥ 光照破坏:置日光灯下照射48 h,取出,加水稀释至刻度,摇匀作为强光破坏供试品溶液。
精密量取上述供试品溶液各20 μL,进样测定,对比未破坏供试品溶液色谱图,酸破坏检出8 个杂质,其中杂质A 含量较高(10.18%);碱破坏检出9 个杂质,其中杂质A 含量较高(0.29%);氧化破坏产生杂质较多;高温破坏、光照破坏未产生新杂质。结果见图3。
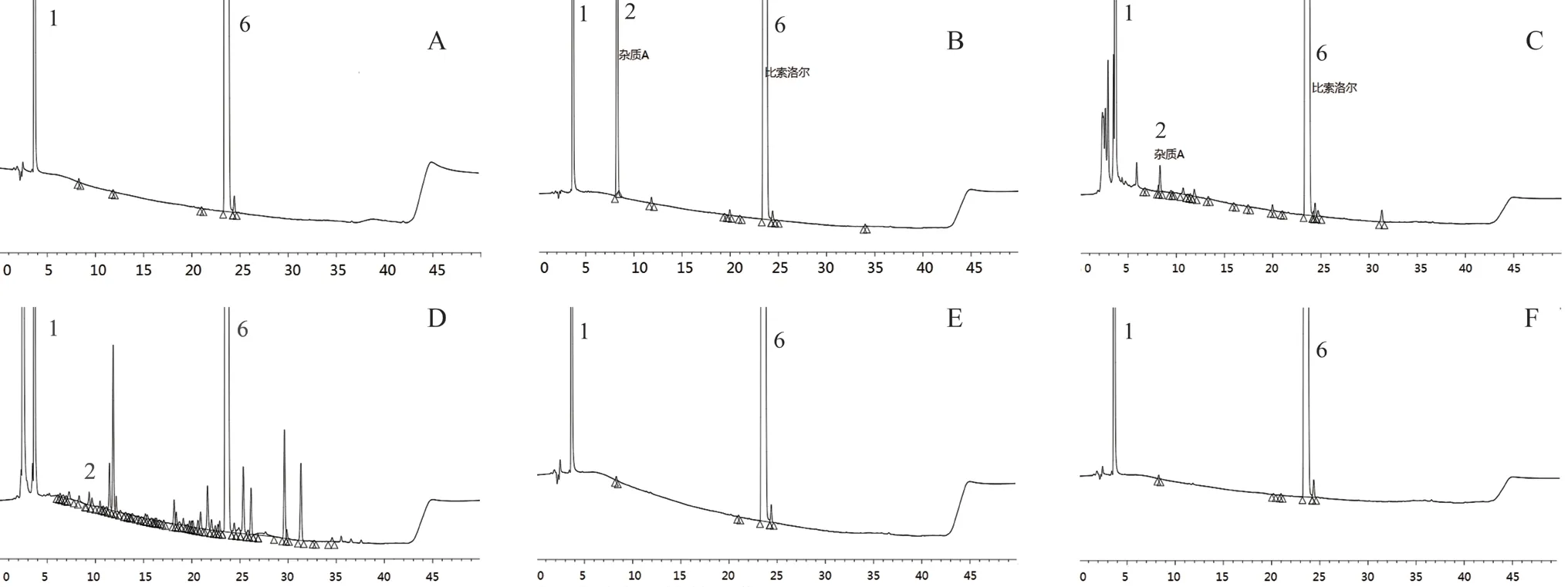
图3 专属性试验色谱图Fig 3 HPLC chromatogram of related substances in bisoprolol fumarate from specificity test
2.3.3 线性关系考察 精密称取杂质A、G、K、L、M、N-醛、羟乙酯、酸及富马酸比索洛尔对照品各约2.5 mg,分别置10 mL 量瓶中,加溶剂溶解并稀释至刻度,摇匀,分别精密量取各溶液适量置同一10 mL 量瓶中,加溶剂稀释制成每1 mL 中分别约含富马酸比索洛尔15 μg、杂质A 15 μg、杂质G 25 μg、杂质K 25 μg、杂质L 25 μg、杂质M 25 μg、杂质N-醛 25 μg、杂质羟乙酯20 μg、杂质酸20 μg 的溶液,作为混合对照品储备液,取适量稀释成限度浓度的1000%、500%、200%、100%、50%、20%的线性溶液。分别精密量取20 μL,照“2.1”项下色谱条件测定,结果见表2。
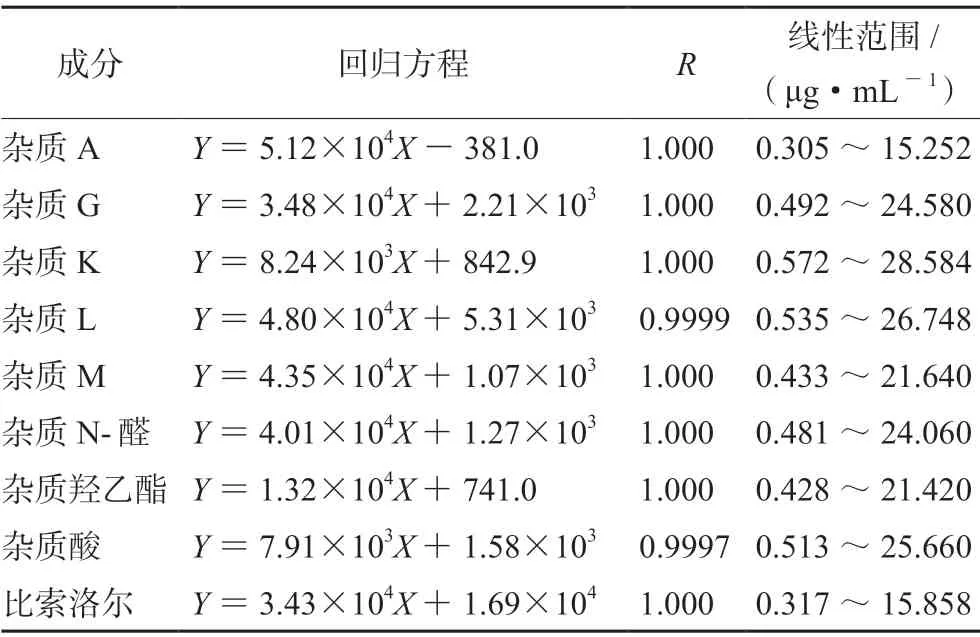
表2 已知杂质线性范围Tab 2 Linearity of the known impurity
2.3.4 精密度试验 取“2.3.3”项下100%质量浓度线性溶液20 μL,按“2.1”项下色谱条件连续进样6 次,结果比索洛尔及各杂质峰面积的RSD均小于1.2%(n=6),表明该方法精密度良好。
2.3.5 检测限与定量限考察 取“2.3.3”项下富马酸比索洛尔及各杂质的对照品溶液,逐级稀释,按“2.1”项下色谱条件进样分析。以信噪比为3∶1,计算检测限,以信噪比为10∶1计算定量限。结果,杂质A、G、K、L、M、N-醛、羟乙酯、酸及比索洛尔的检测限分别为0.031、0.049、0.057、0.054、0.043、0.048、0.043、0.051、0.032 μg·mL-1,定量限分别为0.152、0.246、0.286、0.268、0.216、0.241、0.214、0.257、0.159 μg·mL-1。
2.3.6 稳定性试验 取“2.3.3”项下100%质量浓度线性溶液及“2.2.2”项下供试品溶液,分别于5℃放置0、6、12、17、24 h 后按“2.1”项下色谱条件进样分析,结果混合对照品和供试品溶液在5℃下放置24 h 内杂质个数及杂质量均无明显变化。
2.3.7 加样回收试验 精密称取片剂细粉和胶囊剂内容物细粉适量(本底中含0.152 μg·mg-1的杂质A 及1.562 μg·mg-1的杂质G),共9 份,分别置10 mL 量瓶中,分别精密量取“2.3.3”项下混合对照 品储备液0.5、1.0、1.5 mL 各3 份,置上述量瓶中,加溶剂溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液,按“2.1”项下色谱条件测定,计算回收率。结果杂质A 回收率在100.74%~104.86%,RSD为1.3%;杂质G 回收率在98.15%~106.21%,RSD为2.5%;杂质K 回收率在100.44%~105.23%,RSD为1.6%;杂质L 回收率在104.68%~108.82%,RSD为1.2%;杂质M回收率在100.61%~105.63%,RSD为1.5%;杂质N-醛回收率在98.80%~103.92%,RSD为1.5%;杂质羟乙酯回收率在100.13%~104.44%,RSD为1.3%;杂质酸回收率在90.45%~94.42%,RSD为1.4%。
2.3.8 耐用性试验 分别调整各色谱参数:流速[(1.0±0.05)mL·min-1]、柱温(35℃、40℃)、流动相pH 值(2.5±0.1),取系统适用性溶液注入色谱仪,记录色谱图,各条件下8 个已知杂质与主峰之间的分离度均能不低于1.2,结果重现性较好,方法耐用性良好。
2.3.9 样品测定 分别取各生产企业的原料药或制剂样品适量,按“2.2.2”项下方法制备供试品溶液,按“2.1”项下色谱条件进样分析,结果见表3。
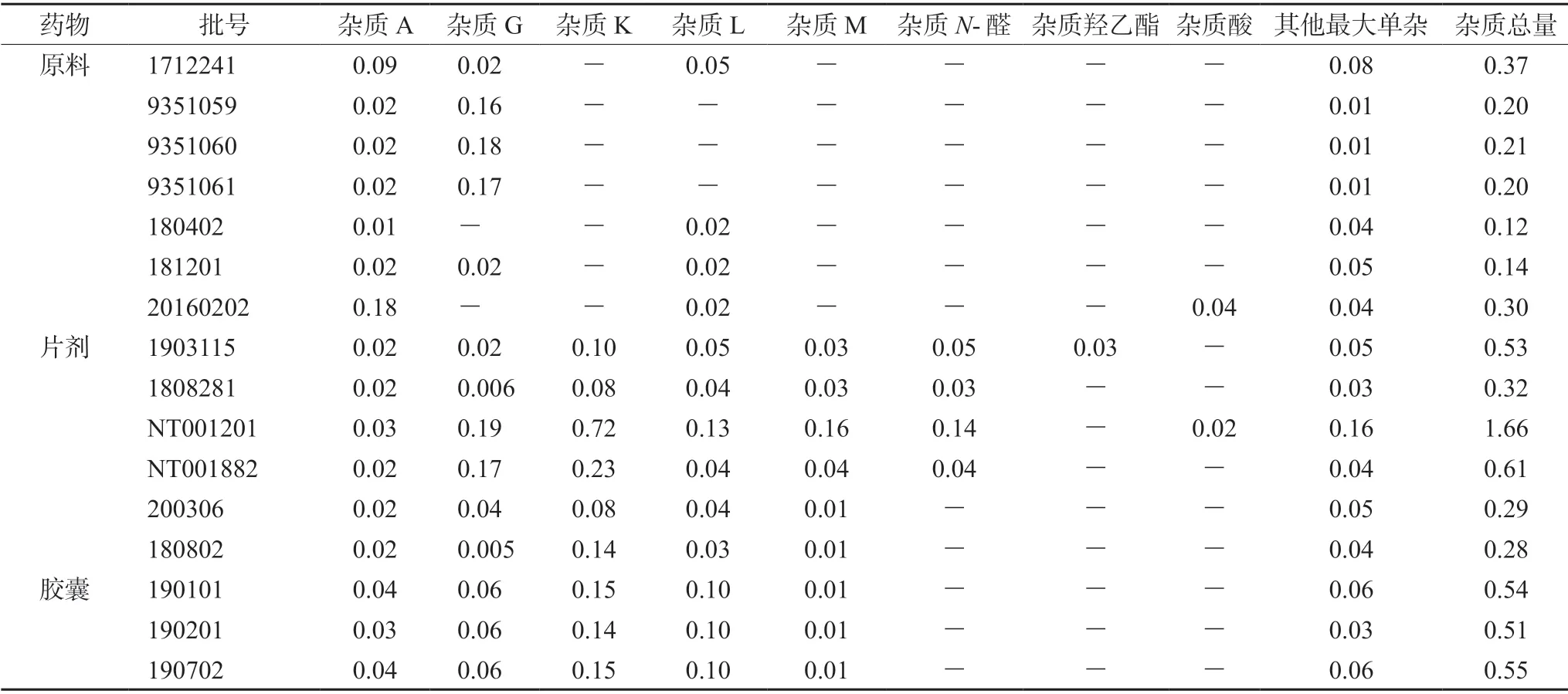
表3 样品中各杂质含量检测结果(%)Tab 3 Impurity determination in the samples(%)
3 讨论
3.1 色谱条件的筛选与确定
BP2020 中收载的已知杂质最多,故本次研究大部分杂质沿用了BP2020[8]中的杂质命名,杂质酸、杂质羟乙酯和杂质N-醛沿用的是进口注册标准JX20150419[9]。收集目前标准中收载的共10 个已知杂质对照品进行系统适用性研究,在参照目前获批的4 个片剂最新国家药品标准(分别沿用的ChP2020 原料有关物质检测系统和进口注册标准JX20150419 有关物质检测系统)和1 个原料最新药品标准[10]的基础上,对色谱条件进行了筛选。结果表明,进口注册标准JX20150419 的色谱系统优化后可以在一个色谱系统中实现10 个已知杂质的良好分离,色谱图见图4。
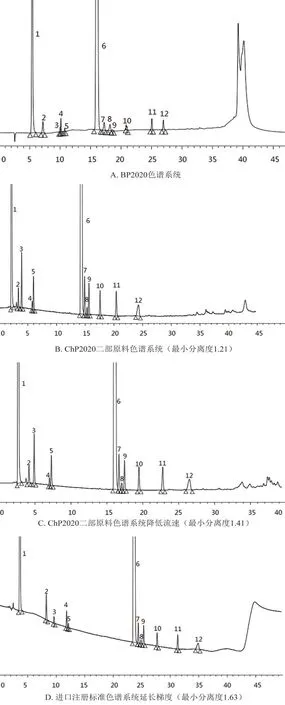
图4 系统适用性试验色谱图Fig 4 System suitability chromatogram of bisoprolol fumarate
3.2 已知杂质的收载
对比国内外标准表明:BP2020 富马酸比索洛尔原料中收载了15 个已知杂质,明确控制了3 个已知杂质;BP2020 片剂中收载并明确控制了6 个已知杂质;片剂进口注册标准JX2015019 中明确控制了7 个已知杂质;片剂标准YBH06092019 及YBH07222020 中收载并明确控制了9 个已知杂质;片剂标准YBH05892018 中收载并明确控制了3 个已知杂质。各标准收载的已知杂质多有交叉,且基本在BP2020 中有收载。结合本品的实际处方及生产工艺分析,本研究初期对有关物质中需要重点控制的8个已知杂质进行了研究。但由于各杂质的特性各异,研究中未找到合适的破坏条件制备同时含多个杂质的系统适用性溶液,考虑到后期标准执行中各杂质对照品的获取问题,在最后的拟订标准中收载了在样品普遍存在且最能反映样品质量的4 个杂质(杂质A、杂质L、杂质G、杂质K)作为已知杂质控制,其余各杂质均作为“其他杂质”一并控制。
3.3 杂质限度的拟订
参考人用药品注册技术要求国际协调会议(ICH)的要求[11-12],拟订原料限度如下:除富马酸及富马酸之前的色谱峰不计外,如有与杂质A、杂质L、杂质G、杂质K 保留时间一致的色谱峰,按外标法以峰面积计算,分别不得过0.3%、0.1%、0.1%、0.1%,其他单个杂质峰面积不得大于对照溶液中比索洛尔峰面积的0.1 倍(0.10%),杂质总量不得过0.5%;拟订制剂限度如下:除富马酸及富马酸之前的色谱峰不计外,如有与杂质A、杂质L、杂质G、杂质K 保留时间一致的色谱峰,按外标法以峰面积计算,分别不得过0.3%、0.5%、0.5%、0.5%,其他单个杂质峰面积不得大于对照溶液中比索洛尔峰面积的0.2 倍(0.2%),杂质总量不得过2.0%。
3.4 影响因素试验
参照ChP2020 四部指导原则<9001>《原料药物与制剂稳定性试验指导原则》,考察3 家企业富马酸比索洛尔片(各1 批)的杂质变化情况。在高温下放置10 d 后,3 家企业的杂质总量均明显增大(A 企业0.19%→0.71%,B 企业1.17%→2.21%,C 企业0.30%→1.15%),但已知杂质变化情况各异(A 企业主要是杂质L 由0.05%→0.19%,B 企业主要是杂质K 由0.42%→1.00%,C 企业主要是杂质L 由0.05%→0.15%)。在高湿下放置10 d 后,A 企业和C 企业样品的杂质总量均无明显变化,但B 企业的杂质总量明显增大(1.17%→1.95%),主要是由于杂质L 和杂质G 的增大引起的。在光照下放置10 d 后,A 企业和C 企业样品的杂质总量均无明显变化,但B 企业的杂质总量明显增大(1.17%→2.03%),主要是由于杂质L、杂质G、杂质M 和杂质N-醛的增大引起的。各企业样品在影响因素试验中表现差异较大。
综上所述,本次研究为富马酸比索洛尔原料、片剂及胶囊提供了一个科学、合理及全面的有关物质分析方法,可以为该品种的系统质量控制提供参考。
猜你喜欢
皮肤病与性病(2021年3期)2021-07-30
皮肤病与性病(2021年3期)2021-07-30
中华养生保健(2020年5期)2020-11-16
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06
数学小灵通(1-2年级)(2017年9期)2017-10-13
学苑创造·B版(2017年1期)2017-02-21
小天使·二年级语数英综合(2016年9期)2016-05-14
爱你(2015年21期)2015-11-15
中国当代医药(2015年36期)2015-03-11
世界热带农业信息(2014年6期)2014-09-12
