
多糖的增稠、胶凝及乳化特性研究进展
2021-08-31 03:30:28李秀秀郭玉蓉
食品科学 2021年15期
李秀秀,尚 静,杨 曦,薛 佳,2,3,4,*,郭玉蓉,3,4
(1.陕西师范大学食品工程与营养科学学院,陕西 西安 710119;2.农业农村部都市农业重点实验室,上海 200240;3.国家苹果加工技术研发专业中心,陕西 西安 710119;4.西部果品资源高值利用教育部工程研究中心,陕西 西安 710119)
多糖广泛存在于自然界中,是最为丰富的天然高分子资源之一[1]。多糖通常是指由多个单糖分子经失水、 缩合而形成的结构复杂且分子质量庞大的一类物质。根据来源及结构特点,可将多糖划分为植物多糖、 动物多糖、微生物多糖、线性结构多糖、分支化多糖、中性电荷多糖、阴离子多糖、阳离子多糖、亲水性多糖、表面活性多糖等[2]。虽然多糖种类繁多,结构差异较大,但从广义层面而言,凡是符合高分子化合物概念的碳水化合物及其衍生物均可称为多糖。其中,食品多糖特指食品加工过程中被允许作为添加性成分,用以提高食品感官、质构特性及营养价值的多糖,是食品胶的重要组成部分。因此,食品多糖的范围相对较窄,某种程度上特指已经商业化的多糖,如果胶[3-4]、结冷胶[5]、琼脂糖[6]、黄原胶、卡拉胶[7]、淀粉及改性淀粉[8]、纤维素及其衍生物等[2]。除淀粉外,大多数食品多糖不能被人体的消化酶水解,但这些多糖普遍具有膳食纤维的功能价值,可起到降血糖、降血压、降低胆固醇以及改善人体肠道菌群平衡的作用[9-10]。
多糖本质上属于天然高分子,因此具有高分子化合物的一般属性。例如,当多糖完全水化后,其水溶液常表现出各异的流变学特性,包括增稠、胶凝及乳化特性等。一般地,多糖溶液的流变学特性受多糖自身分子结构和外界环境因素的影响。其中,影响多糖流变学特性的结构参数主要包括分子质量、电荷密度、分支化程度等因素,外界环境因素则主要包括pH值、温度、离子类型和强度、有无其他共溶质存在等。虽然影响多糖流变学特性的因素众多,且影响途径不同,但这些因素影响多糖溶液流变学特性的本质在于影响多糖在水溶液中的超分子结构[11-12]。对于典型的凝胶多糖,如结冷胶、卡拉胶、琼脂糖等,当溶液体系由溶胶态向凝胶态转变时,多糖分子彼此间往往形成稳定的交联区,构成空间三维网络结构[13-14]。当体系由凝胶态向溶胶态转变时,稳定的分子间交联重新打开,体系再次以溶胶形式存在,因此这类多糖常常表现出可逆的凝胶化过程。对于非凝胶多糖,如黄原胶、魔芋胶、刺槐豆胶等[15-17],水化后多糖分子倾向于形成非稳定的分子间聚集,这样的结构容易被外界应力扰乱而解聚集,因此这类多糖形成的溶液具有良好的增稠效果,但胶凝能力较差。此外,一些多糖分子结构中含有少量的疏水官能团,使得它们同时表现出亲水和疏水的特性,称为表面活性多糖或两亲性多糖。表面活性多糖能够吸附至油-水界面,并具有在油滴表面形成一层水化膜、防止油滴聚集的能力,因此表面活性多糖可作为乳化剂制备乳液[8,15,18-20]。
食品多糖的胶凝、增稠、乳化等流变特性和分子结构密切相关,本质上是多糖超分子结构的宏观体现[3,20-26]。 因此,理解食品多糖流变学特性的本质需要从解析多糖超分子结构的角度入手。虽然目前国内外有关多糖资源开发利用的文献已有不少报道[27-30],但鲜有从多糖高分子化合物的本质属性出发形成专门解读多糖流变学特性的文献综述。另一方面,随着国内食品工业的快速发展,越来越多的食品多糖被用于改善和优化食品结构,因此对食品多糖流变学本质进行解读显得尤为必要。因此,本文探讨了食品多糖胶凝、增稠、乳化等流变学特性及其超分子结构之间的相互关联,总结了影响多糖流变学特性的一般规律,旨在为多糖在食品工业中的进一步应用奠定理论依据。
1 多糖溶液特性
1.1 多糖稀溶液特性
当多糖完全水化后,以溶液形式存在。根据临界接触浓度c*的概念,多糖溶液一般可分为稀溶液和半稀溶液两个区域。c*表示多糖分子在溶液中相互接触的临界浓度[16,31]。当多糖浓度低于c*时,多糖以单一分子存在,彼此不产生接触和交联;当浓度高于c*时,多糖分子开始发生彼此接触甚至互相穿插、挤压。因此,当多糖浓度越过c*时,溶液黏度特性将发生突变。通常,多糖溶液的c*和多糖分子在溶液中的流体力学体积有密切关系,流体力学体积越大,c*越小。由于特性黏度η是描述聚合物分子流体力学体积大小的度量,因此测定多糖溶液的η值可以估计流体力学体积。通常,不同类型的多糖分子质量、分支化程度以及分子构象均不同,因此它们的η值差异也较大。即便对于同一种多糖,当溶液所处的外界环境因素(如温度、pH值、离子强度等)不同时,其分子构象不同,因此η值也不同。理论上,多糖的η值和c*具有反比关系,可以近似表示为c*=α/η,其中,α为常数,其值大小一般为4。显然,这一经验式为预测多糖溶液c*的简单算法,但c*也可进行实际测定。
根据文献[16]中不同质量分数(w)多糖溶液的增比黏度(ηsp)绘制图1。lgηsp和lgw的关系可由两个线性区表示:在多糖质量浓度较低时,斜率约为1.4;在多糖浓度较高时,lgηsp和lgw线性关系曲线的斜率约为3.3,两个线性区的交点即为c*。当多糖浓度低于c*时,多糖分子彼此不接触,此时多糖分子和水分子的摩擦力成为影响多糖溶液黏度的主要因素,当浓度高于c*时,多糖分子彼此接触,产生较大的摩擦,此时多糖分子和水分子之间的摩擦、多糖分子彼此之间的摩擦共同决定溶液的黏度。因此,当多糖浓度高于c*时,溶液黏度明显增大。然而,需要注意的是c*=α/η[32]这一经验式仅适用于无规卷曲构象的多糖[32],即多糖分子彼此之间不发生聚集。但在一些情况中,多糖在溶液中常发生分子间聚集,导致实际测定的c*和采用经验式推导的c*存在明显偏差。这是因为当溶液中多糖分子彼此聚集形成分子间聚集体时,测定的η增大,由于c*和η为反比关系,使得实际测定的c*低于预测的c*。因此,通过对比预测的c*和实际测定的c*,可以间接推测出溶液中多糖 分子是否发生了聚集。即当实际测定的c*低于理论推测的c*时,表明发生了分子间聚集,反之则说明不存在分子间聚集[31]。
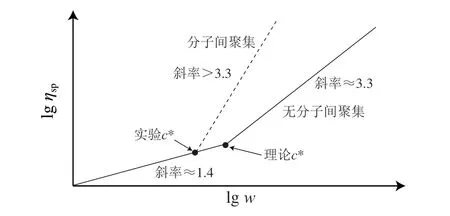
图1 多糖溶液浓度与其增比黏度的关系[16]Fig.1 Relationship between the concentration of polysaccharide solution and its specific viscosity[16]
1.2 多糖浓溶液特性
当多糖浓度低于c*时,溶液中仅存在多糖分子和水分子之间的摩擦、水分子彼此之间的摩擦,两者之和决定了溶液的黏度。通常,水分子之间的摩擦力很低,可忽略不计,因此多糖溶液的黏度主要取决于多糖分子和水分子之间的摩擦。此时,多糖溶液近似表现出牛顿流体的特性。当多糖的浓度高于c*时,多糖分子彼此接触,导致多糖分子之间的摩擦力急剧增加,具体表现为溶液黏度显著增加。此时,溶液的剪切稀化现象逐渐凸显。由于不同结构特点的多糖在水溶液中的超分子结构不同,因此剪切稀化行为也不同,通常采用表观黏度随剪切速率的变化关系来描述多糖的剪切稀化效应。当剪切速率较低时,多糖分子聚集结构被外界剪切应力打破的速率和结构重建的速率几乎相等,因此表观黏度不随剪切速率的增加而变化,此时溶液的特性黏度和零剪切黏度相等,可由η0表示。当剪切速率继续增加时,多糖聚集结构的破坏速率大于重建速率,表观黏度呈现降低趋势,剪切速率越大,表观黏度越低,这一阶段即为剪切稀化区。当剪切速率继续增加时,多糖聚集结构的破坏速率远远大于重建速率甚至多糖分子沿着剪切应力的方向发生定向重排,此时溶液的表观黏度也不随剪切速率的增加而增加,整体维持在较低的黏度水平(由η∞表示)。由于不同类型的多糖具有不同的分子结构,因此它们的流体力学体积不同,溶液中的超分子结构不同,因此它们的特性黏度变化趋势也不同。
此外,当多糖浓度增加至c*以上时,多糖分子彼此接触并互相挤压,降低了单个多糖分子的流动性,因此当外界剪切应力打破溶液中多糖的超分子结构后,多糖分子总是需要一定的时间重新恢复聚集结构,浓度越大,多糖分子挤压程度越高,恢复结构所需要的时间就越长,因此剪切稀化效应发生时所对应的剪切速率向更低的数值移动,根据文献[32]数据绘制高浓度多糖溶液与低浓度多糖溶液剪切黏度变化如图2所示。

图2 高浓度多糖溶液与低浓度多糖溶液剪切黏度变化[32]Fig.2 Shearing viscosity of polysaccharide solutions at high and low concentrations[32]
由于不同类型的多糖具有不同的η0,且同一类型的多糖在不同的浓度条件下也具有不同的η0,因此直接比较多糖溶液的黏度特征非常困难[33]。为了得到不同类型多糖剪切黏度的一般性规律,Morris等[32]定义多糖溶液特性黏度η降低至零剪切黏度η0十分之一时对应的剪切速率为“剪切稀化参数”,表示为γ0.1。如图3所示,由不同浓度条件下瓜尔豆胶、L(lambda)型卡拉胶、刺槐豆胶、海藻酸钠及透明质酸的归一化结果可知,即便是多糖种类不同、分子质量不同、浓度不同,仍然可以获得重合度较高的归一化曲线,区别仅在于它们的η0和γ0.1不同。此外,Morris等[32]还发现,这一规律对溶液中发生明显分子聚集行为的多糖也同样适用。如图3所示,刺槐豆胶、瓜尔豆胶、海藻酸钠等均存在明显的分子聚集行为,但仍然可以归一化。原因可能是,虽然这些多糖在溶液中能够发生分子聚集,但分子聚集所需要的时间远远超过多糖分子间彼此物理碰撞和接触所需要的时间,因此在剪切作用下,分子聚集行为对溶液黏度的影响几乎可以忽略不计。
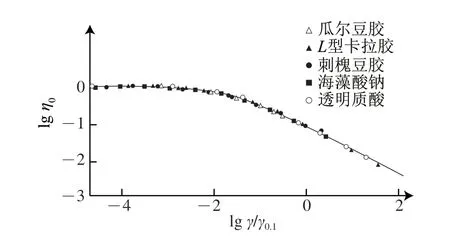
图3 归一化后多糖溶液黏度变化曲线[32]Fig.3 Generalized shear thinning behavior of concentrated solutions of disordered polysaccharides[32]
2 不同因素对多糖凝胶化的影响
当多糖超过某一临界浓度时,有的多糖能在外界环境因素的诱导下发生凝胶化,称为凝胶多糖,对应的临界浓度称为临界胶凝浓度。然而,有关凝胶的定义却一直没有达成一致,主要原因在于评估多糖溶液体系是否形成凝胶的方法多种多样,因此凝胶的定义也不尽相同。其中,最为简单的方法是试管倒置法。当倒置试管时, 多糖体系不从试管底部流出,即可认为形成了凝胶。然而,流变学中,凝胶定义为储能模量G′高于损耗模量G”的软物质体系,只要符合这一标准,均可以从流变学角度判定为凝胶[34]。但某些凝胶体系由于结构较弱,虽然符合G′>G”,但并不符合试管倒置法的判别标准,通常将这样的体系归于弱凝胶的范畴。对于凝胶结构较强的多糖体系,G′应该比G”高一个数量级,且在较宽泛的振荡频率范围内(如0.1~100 rad/s)G′和G”保持恒定,不随振荡频率的增加而变化。目前,这一标准成为了食品流变学领域中评估食品多糖凝胶的主要依据,也排除了“弱凝胶”和“流体凝胶”的范畴。当多糖完全水化形成溶胶后,有的多糖体系能够在改变外界环境因素的条件下形成凝胶,有的则不能。前者通常称之为凝胶多糖,后者定义为非凝胶多糖。两者最大的区别在于,凝胶多糖可以在水溶液中形成稳定的分子间交联,构成空间三维网络结构,但非凝胶多糖不能形成稳定的分子间交联,因此在外界应力的作用下,非凝胶多糖的超分子结构很容易被打破,外观表现为体系具有流动性。
图4为两种典型的多糖凝胶化机制[35]。对于凝胶多糖,凝胶化需要在合适的外界条件下发生,如适宜的温度、pH值、离子浓度、多糖浓度等。对于负电荷密度较高的多糖,如海藻酸钠和低酯果胶等,分子结构中含有较多数目的羧基,因此对二价离子(如Ca2+)具有较强的结合能力,通常一个钙离子可以结合两个解离的羧基基团[36-38]。当存在钙离子时,海藻酸钠和低酯果胶可以与钙离子形成分子间交联,构成稳定的空间三维网络结构(图4A)。这一凝胶化机制通常被描述为“蛋-盒模型”[39-42]。但对于某些食品多糖,它们本身具有良好的胶凝能力,可以在不添加钙离子或其他阳离子的条件下形成凝胶。如图4B所示,这类多糖往往电荷密度很低,凝胶化过程更多地依赖于分子螺旋聚集体的形成,主要包括结冷胶、琼脂糖、卡拉胶等。然而,即便是对于这类凝胶多糖,添加阳离子也可以明显促进凝胶化过程。一方面,添加阳离子屏蔽了多糖分子链段上的负电荷,降低了多糖分子间的静电斥力,有利于螺旋聚集体的形成;另一方面,添加阳离子改变了水溶液的极性环境,降低了多糖的溶解度(盐析效应),同样也利于多糖分子彼此聚集。不同多糖凝胶的流变学特性如表1所示。

图4 两种典型的多糖凝胶化机制[35]Fig.4 Two typical gelling mechanisms of polysaccharides[35]

表1 不同多糖凝胶的流变学特性Table 1 Rheological properties of different polysaccharide gels
2.1 分子结构对多糖凝胶化的影响
即便是对于同一种多糖,分子结构的不同也会引起凝胶性质的较大差异。一般而言,多糖分子质量越大,越有利于形成凝胶。此外,对于海藻酸钠和低酯果胶这类依靠钙离子存在而发生凝胶化的多糖,分子链段上羧基的分布模式也是影响与钙离子结合的重要因素。通常, 当羧基随机分布于多糖链段时,胶凝能力较弱,当羧基连续分布于多糖链段时(“区域”分布模式),更有利于形成凝胶。有研究表明,当连续6 个解离的羧基和钙离子结合时,相邻的果胶分子才能形成稳定的交联区,显然,“区域”分布模式更利于多糖分子与钙离子结合形成稳定交联区[3]。多糖分支化程度也对多糖的胶凝能力有一定影响。在同等分子质量的前提下,分支化程度越高的多糖,流体力学体积越低,因此不利于形成浓溶液,一定程度上降低了多糖的胶凝能力。尤其是对于低酯果胶而言,分支化程度越高,分子间空间位阻越大,越影响果胶分子“蛋-盒”模型的形成。然而,近年来也有报道指出,分支化程度较高的果胶具有更好的胶凝能力,这是因为侧链分支相互缠绕,有助于凝胶网络的形成。
此外,某些关键基团的含量也会对多糖的凝胶化过程和凝胶特性产生明显影响,例如果胶、结冷胶及卡拉胶等。高酯果胶和低酯果胶最主要的区别是酯化程度不同,但足以使两者具有不同的胶凝机制[3]。低酯果胶凝胶化需要钙离子参与,凝胶可以在相对较宽的pH值范围(如pH 3.5~9.5)内形成[36],但高酯果胶凝胶化不需要钙离子参与。结冷胶分为高酰基和去酰基两种形式,微生物发酵后直接产生的结冷胶为高酰基形式,通过热碱处理除去高酰基结冷胶分子链上的酰基基团后可得到去酰基结冷胶[50]。去酰基结冷胶的胶凝机制一般解释为:高温条件下,结冷胶以无规卷曲构象存在,随溶液温度降低,结冷胶分子逐渐形成双螺旋结构,当温度进一步降低时,双螺旋彼此聚集形成螺旋聚集体,引起空间三维网络形成,导致凝胶化[51]。加热时,结冷胶凝胶逐渐融化,但凝胶融化温度通常高于60 ℃,呈现明显的热滞后效应。高酰基结冷胶的酰基取代基分为乙酰基和甘油酰基两种,据报道显示,乙酰基被认为是阻碍高酰基结冷胶分子螺旋聚集体形成的关键因素,而甘油酰基主要起到稳定高酰基结冷胶双螺旋的作用[52]。因此,高酰基结冷胶凝胶形成过程和凝胶融化过程中均不涉及螺旋聚集体的形成和解螺旋,所以高酰基结冷胶没有明显的热滞后效应,凝胶化温度和凝胶融化温度几乎一致[53]。类似的例子还包括卡拉胶,卡拉胶主要包括K型(kappa)、I型(iota)、L型3 种形式,区别仅在于硫酸酯基含量和连接位点不同,但足以引起它们在胶凝特性方面的差异[54-58]。例如,K型卡拉胶对钾离子非常敏感,但对钠离子不敏感,在K+存在的前提下,K型卡拉胶的凝胶化过程被极大促进,形成强度较高、质地硬而脆的凝胶。L型卡拉胶对一价阳离子和二价阳离子均不敏感。I型卡拉胶可以和钙离子结合形成柔软富有弹性的凝胶。
2.2 外界因素对多糖凝胶化的影响
凝胶化的本质是多糖水溶液在适当的外界刺激下,多糖分子发生聚集,形成稳定的分子间聚集区,从而形成 三维网络结构的过程。通常,对于冷致型凝胶多糖,降低温度可以促使多糖溶液从溶胶态转变为凝胶态。对于疏水基团含量较高的多糖,疏水作用力是诱导发生凝胶化的主要因素[2]。当加热这类多糖的水溶液至凝胶化温度以上时,凝胶才能形成,称之为热致凝胶。例如,甲基纤维素(methylcellulose,MC)和羟丙基甲基纤维素(hydroxypropyl methyl cellulose,HPMC)均是典型的热致型凝胶多糖。有报道显示,MC的凝胶化温度约为52 ℃,HPMC的凝胶化温度范围较宽,约为63~80 ℃[2]。当冷却MC或HPMC的凝胶时,分子间疏水作用力逐渐减弱,导致凝胶网络解聚集,促使多糖分子从凝胶状态重新转变为溶液状态。pH值对多糖凝胶化的影响在于改变带电荷多糖的分子构象以及分子间相互作用力。因此,改变溶液pH值可以诱导一些多糖发生凝胶化,或促进凝胶形成。例如,据报道显示,结冷胶溶液的pH值降低至3.5时,凝胶强度可增加4 倍以上[59]。此外,pH值也是影响海藻酸钠凝胶化的重要因素,海藻酸钠电荷密度较高,在不含二价阳离子的情况下,溶液一般以黏稠流体的形式而存在。然而,当pH值缓慢降低至3.0时,海藻酸钠可形成凝胶,原因在于降低溶液pH值后,减少了海藻酸钠分子间的静电斥力,有利于分子聚集。阳离子对多糖凝胶化的影响分为3 个方面:1)一价阳离子通过遮蔽多糖分子链上的负电荷降低分子间静电斥力,促进分子间聚集;2)二价或多价阳离子直接和多糖分子中解离的羧基结合形成分子间交联;3)阳离子浓度过高时,改变了溶液的极性环境,可能引起多糖分支的“盐析”效应。
此外,多糖的凝胶化过程也受其他因素的影响。例如,在少量蔗糖存在的情况下,低酯果胶的凝胶强度明显提升,一方面,蔗糖作为脱水剂,破坏了果胶分子表面的水化膜,有利于果胶分子彼此靠近与钙离子形成交联;另一方面,蔗糖本身也可以作为填充成分加强果胶分子的凝胶网络。
3 非凝胶多糖
与凝胶多糖相比,非凝胶多糖水化后以流体形式存在,因此在食品领域中主要用作增稠剂、分散剂、乳化剂等。需要注意的是,所有的凝胶多糖均具有增稠特性,这是由它们的高分子本质决定的。例如,当凝胶多糖在水溶液中的浓度较低,不足以形成较强的三维凝胶网络时,溶液宏观上表现为黏稠体系,和非凝胶多糖相似。某些多糖,如黄原胶、瓜尔豆胶、魔芋胶等,自身可以形成微弱的分子间聚集,使得它们的溶液常表现出类似于弱凝胶的特点,即G′>G”,但这样的结构容易被剪切应力破坏而产生流动性。因此,这类多糖仍然归属于非凝胶多糖。
食品工业中,非凝胶多糖主要用作增稠剂。由于不同结构特点的多糖在溶液中的超分子结构不同,因此溶液黏度特征也不同。除上述总结的多糖溶液特性外,剪切稀化行为是评估非凝胶多糖流变特性的另一重要因素,通常采用表观黏度随剪切速率的变化关系来描述。通常,不同结构特点的多糖剪切稀化行为呈现较大差异。例如,黄原胶一般具有最为显著的剪切稀化效应,低剪切速率条件下,黄原胶溶液表观黏度很高,随剪切速率增加,表观黏度急剧降低,使得黄原胶具有最为明显的剪切稀化效应。其他具有显著剪切稀化效应的多糖主要为葡甘露聚糖类。主要原因是,黄原胶和葡甘露聚糖均能发生微弱的分子间聚集,形成类似于“弱凝胶”的结构,因此低剪切速率时,溶液表观黏度很高,但这种结构容易被剪切应力破坏,所以高剪切速率时,溶液黏度急剧降低[60-63]。具有显著剪切稀化效应的非凝胶多糖一般作为性能优良的增稠剂或稳定剂广泛应用于饮料工业中。
同样地,非凝胶多糖的增稠特性也受到外界环境因素,如温度、pH值、盐离子等的影响。一方面,这些因素可以影响非凝胶多糖的分子构象,进一步影响多糖的流体力学体积;另一方面,这些因素也可以改变多糖分子间作用力,促进多糖分子聚集,形成微弱的聚集网络,增加溶液黏度。例如,对于含有羧基的非凝胶多糖,随pH值增加,多糖分子的电荷密度也增加,分子间静电斥力增大,多糖分子扩张,流体力学体积增大,有利于增加溶液黏度。然而,加入阳离子时,多糖分子携带的负电荷被屏蔽,降低了分子间静电斥力。但在某些特殊情况中,当多糖分子携带的电荷被屏蔽后,多糖可能发生分子间聚集,形成微弱的三维网络结构,反而增加了多糖溶液黏度。当温度升高时,对于以分子间氢键为主要作用力的多糖,其溶液黏度往往呈现出降低趋势,但对于以疏水作用为主要结合力的多糖而言,溶液黏度往往表现出明显的增加,甚至导致凝胶化。
需要注意的是,非凝胶多糖和凝胶多糖的区分并没有严格的界限,而是根据多糖在食品工业的常规用途而进行的划分。在一些特殊的条件下,某些非凝胶多糖也可能发生凝胶化。例如,有报道显示,黄原胶在冻融条件下可以发生凝胶化[53]。同样地,当严格控制使用条件时,某些凝胶多糖也可能不发生凝胶化。
4 表面活性多糖
4.1 表面活性多糖的界面吸附特性
区别于小分子表面活性剂,表面活性多糖的表面活性特指多糖分子吸附在油-水界面的能力,而小分子表面活性剂的表面活性则指降低油-水界面张力的特性。多糖的 表面活性构成了其作为乳化剂的基础[21]。通常,多糖乳化剂降低油-水界面张力的能力非常有限,因此多糖的乳化能力主要来源于两个方面:1)吸附在油-水界面的能力;2)在乳滴表面形成一层水化膜,防止油滴聚集的能力[19]。一旦乳液形成,需要多糖分子永久性地吸附在油-水界面,提供保护作用,这就要求多糖分子必须含有一定数目的疏水基团[64-66]。此外,乳滴表面水化膜的厚度也是影响多糖乳液稳定性重要因素,水化层越厚,乳滴之间的空间位阻作用也越大,乳液稳定性越好。除疏水基团含量外,影响多糖乳液稳定性的因素还包括多糖的电荷密度、分子质量,以及乳液离子强度、温度等[67-68]。近年来,国内外对多糖乳化特性的研究越来越重视,相关报道日益增多,目前国内已有不少文献详细总结了多糖的乳化特性及多糖乳液的稳定策略[69-75]。
4.2 表面活性多糖的分子聚集行为
表面活性多糖的另一个特殊性质是其在水溶液中的分子聚集特性[76-77]。表面活性多糖分子结构中含有一定数目的疏水基团,因此疏水作用力可能会对多糖在溶液中的存在状态造成一定影响。如果疏水作用力足够强烈,多糖分子能够在疏水作用力的作用下形成以疏水微区或胶束为交联的分子间聚集结构[78]。一般地,当表面活性多糖浓度增加到某一临界值时,多糖水溶液中开始形成胶束,这一浓度通常称为临界胶束浓度。多糖的临界胶束浓度概念和小分子表面活性剂的临界胶束浓度概念类似,但由于多糖溶液同时又兼具聚合物溶液的一般特性,使得表面活性多糖的分子聚集行为更加复杂[79]。此外,表面活性多糖属于聚合物表面活性剂的范畴,而聚合物表面活性剂在水溶液中的聚集行为受疏水基团含量和分布模式的影响。有文献报道,根据聚合物表面活性剂疏水官能团的分布模式,可将聚合物表面活性剂分为大分子表面活性剂和聚皂两大类[72]。大分子表面活性剂的结构和小分子表面活性剂结构类似,是由亲水头链和疏水尾链共同构成的聚合物分子。聚皂则指疏水单元和亲水单元嵌段形成的共聚物分子。因此,根据定义来看,几乎所有的天然表面活性多糖和疏水改性后的多糖均属于聚皂。然而,大多数天然多糖所含的疏水基团数目有限,因此形成胶束的能力较弱。目前只有零星研究报道了天然表面活性多糖形成胶束或疏水微区的能力。例如,有研究报道,高酰基结冷胶凝胶中存在明显的疏水微区,且随多糖浓度降低,疏水微区的密度也降低[31]。
5 结 语
多糖种类繁多,结构差别较大,不同的多糖常表现出各异的流变学特性。外界环境因素影响多糖流变学特性的本质在于影响多糖的超分子结构,因此多糖分子 在溶液中的聚集结构是影响多糖流变学特性的根本因素。本文通过综述多糖流变学特性的基本原理,可为多糖在食品工业中的应用提供理论参考。
猜你喜欢
食品与发酵工业(2024年3期)2024-02-22 14:47:44
石油沥青(2021年4期)2021-10-14 08:50:52
山东冶金(2018年5期)2018-11-22 05:12:46
西安建筑科技大学学报(自然科学版)(2016年1期)2016-11-08 12:15:18
合成化学(2015年9期)2016-01-17 08:57:24
合成化学(2015年10期)2016-01-17 08:56:33
铁道科学与工程学报(2015年4期)2015-12-24 12:11:01
华东师范大学学报(自然科学版)(2014年1期)2014-04-16 02:54:58
中国海洋大学学报(自然科学版)(2014年8期)2014-02-28 12:21:27
食品工业科技(2013年10期)2013-08-07 09:13:56
