
NMDA重叠MOG抗体阳性的脑炎2例及文献复习
2021-08-31 01:42马红梅闫鹤立
中风与神经疾病杂志 2021年7期
杨 丹, 马红梅, 闫鹤立
抗NMDA受体(Anti-N-methyl-D-aspartate receptor)脑炎是一种中枢神经系统的自身免疫性疾病,由抗NMDA受体抗体介导。NMDA受体由2个GluN1亚单位和2个GluN2或GluN3亚单位组成,主要分布于海马、前脑及边缘系统[1]。该病临床表现为急性或亚急性起病,以精神行为异常、自主神经功能障碍、癫痫发作、中枢性低通气、不自主运动等为主要表现。MOG抗体病(myelin oligodendrocyte glycoprotein antibody-associated disease,MOG-AD)是由髓鞘少突胶质细胞糖蛋白(MOG)抗体介导的一种特发性中枢神经系统脱髓鞘疾病。MOG是特异性表达于中枢神经系统少突胶质细胞表面的糖蛋白,位于髓鞘结构的最外层,参与髓鞘粘附,维持髓鞘结构完整性,是中枢神经系统脱髓鞘病的靶抗原。在体外诱导补体介导下,MOG 抗体可以破坏少突胶质细胞的微管组织,导致髓鞘碱性蛋白变性以及轴索蛋白表达异常,从而引起中枢神经系统病变。MOG抗体阳性常出现在单时相的急性播散性脑脊髓炎、AQP4(水通道蛋白-4) 抗体阴性的视神经脊髓炎、视神经炎和横贯性脊髓炎中[2]。近年来有文献报道了NMDA脑炎与MOG抗体病重叠的病例,但检索发现报道的病例在临床症状及影像改变差异较大[3,4],临床对其认识不足,容易导致误诊或漏诊。本文中报道了两例以大脑皮质受累为主的MOG和NMDA重叠的患者,并进行文献复习,类似病例报道较少,希望本文能为临床诊疗提供更多的参考。
1 临床资料
1.1 病例1 男,28岁,汉族。主诉“头痛、发热、反应迟钝23 d,发作性意识丧失伴肢体抽动4 h。”于2019年8月18日收入我院。入院23 d前(2019年7月27日)患者开始出现头痛,为全头部胀痛,自觉有发热,未测体温,症状持续存在,自行服用感冒药治疗(具体不详)。随后几天同事发现其出现反应迟钝、言语减少。2019年8月14日患者出现一过性意识丧失,未摔伤,后自行清醒,清醒后不能回忆(具体情况不详)。自行到当地医院行头部CT:左颞脉络膜囊肿?,余未见异常。入院4 h前(2019年8月18日)患者出现意识丧失,摔倒在地,伴双眼上翻、口吐白沫、四肢强直抽动,无舌咬伤,无二便失禁,持续3 min抽动停止,患者意识逐渐恢复,意识清醒后发现其存在表达困难、右侧肢体无力,行走困难,被同事送入我院。既往体健。体格检查:体温38.2 ℃,脉搏68次/min,呼吸20次/min,血压134/78 mmHg,双肺呼吸音粗,余心肺查体阴性。神经系统查体:意识朦胧,反应迟钝,高级皮质功能减退,不完全性混合性失语,双侧瞳孔等大正圆,对光反应灵敏,右侧肢体肌力 5-级,余肢体肌力5级,四肢肌张力、感觉查体正常,共济查体不能配合,右侧病理征阳性,脑膜刺激征阴性,余查体未见明显异常。
入院后辅助检查:①凝血常规、甲功七项、降钙素原、输血八项、糖化血红蛋白、血叶酸、VB12、血G试验、GM 试验、抗核抗体谱、抗中性粒细胞胞浆抗体、男性肿瘤标记物未见异常。血常规+CRP:白细胞:14.39×109/L;C反应蛋白:21 mg/L;中性粒细胞百分比:81.6%;淋巴细胞百分比:12.1%。生化:谷氨酰转肽酶:65 U/L;同型半胱氨酸:18.8 μmol/L。血沉:45 mm/h。②住院期间四次腰穿化验结果(见表1)。特殊化验:血免疫球蛋白正常,脑脊液(CSF)免疫球蛋白:IgA 1.84 mg/dl↑、IgM 0.45 mg/dl↑、IgG 6.27 mg/dl↑。CSF X-pert阴性,血及CSF ADA、结明三项阴性。血及CSF隐球菌荚膜抗原定性阴性。NMDA 受体抗体:CSF 1∶100,血1∶30,抗MOG抗体:CSF 1∶320,血1∶10。血及CSF:AQP-4、抗MBP抗体、副肿瘤相关抗体阴性。血布氏杆菌、莱姆抗体、囊虫抗体阴性。脑脊液病原学(DNA、RNA)二代测序未查到特异性病原体。血肺炎支原体抗体阴性。脑脊液细菌培养阴性。③影像检查:头部MRI双侧额叶、颞叶、顶枕叶皮质异常信号(见图1)。④治疗、预后:入院后给予“左乙拉西坦抗癫痫、阿昔洛韦抗病毒、七叶皂甙钠减轻脑水肿、免疫球蛋白 每日0.4 g/kg 静点(共5 d),甲泼尼龙500 mg静点3 d,250 mg 3 d,160 mg 3 d,120 mg 3 d,80 mg 3 d,后改为泼尼松片60 mg口服,每周减1片;头孢曲松控制肺炎”等治疗,患者仍频繁出现癫痫发作:主要有三种形式:a:发作性右侧或双侧口角抽动。b:双眼向一侧凝视、头向右偏伴右侧肢体抽动。c:全身强直-阵挛发作。我们加用苯巴比妥肌注、丙戊酸钠泵入控制癫痫发作。患者病情仍持续加重,意识障碍加深,右侧肢体肌力明显下降,为0~1级,出现Ⅱ型呼吸衰竭,抗生素升级为美罗培南;因肝功能异常,停阿昔洛韦改为膦甲酸钠抗病毒。2019年9月11日家属拒绝行气管插管,要求自动出院于外院治疗,出院3 d后患者死亡。

图1 病例1影像 A-C:DWI可见双侧颞叶、海马、额叶、顶枕叶皮质广泛高信号;D:T1WI呈低信号;E、F:T2WI呈高信号;G-J:Flair呈高信号,K、L:核磁增强未见病灶明显强化
1.2 病例2 男,27岁。主因“言语不清伴右上肢无力27 d,加重伴肢体抽搐、精神行为异常1 d。”于2020年11月4日收入我院。入院27 d前(2020年10月5日)患者突发言语不清,表现为表达困难伴口齿不清、右侧口角歪斜,同时伴面部及右上肢麻木、无力,持续2 h左右肢体无力部分缓解,伴全头部持续胀痛,无恶心、呕吐,伴反应迟钝,持续存在。无意识丧失、肢体抽搐。当地医院行头部CT未见明显异常。既往体检。体格检查:体温37.1 ℃,脉搏88次/min,呼吸21次/min,血压131/86 mmHg,心肺查体阴性。神经系统查体:反应迟钝,计算力下降,记忆力、定向力、理解力正常,右侧鼻唇沟变浅,言语欠清,伸舌稍右偏,右上肢肌力5-级,脑膜刺激征阴性,余查体未见异常。
辅助检查:2020年10月6日头部MRI可见左额颞顶叶脑皮质异常信号,增强未见明显强化(见图2)。2020年10月7日腰穿一般情况(见表2):血及脑脊液自身免疫性脑炎抗体阴性,脑脊液OB、SOB弱阳性,血阴性。脑脊液免疫球蛋白IgA 0.66 mg/dl↑、IgG、IgM阴性,血免疫球蛋白阴性。血及尿线粒体脑病基因筛查阴性。脑脊液病原学(DNA、RNA)二代测序阴性。脑电图:轻度异常。住院期间再次复查头部MRI(2020年10月13日)可见左侧额叶、顶叶异常信号较前增加(见图3)。初诊为“病毒性脑炎”,给予“阿昔洛韦”静点21 d等治疗,2020年10月26日患者上述症状基本缓解,复查腰穿一般情况(见表2),神经系统查体:反应稍慢,余无异常。2020年10月26日带口服阿昔洛韦片出院。出院3 d(2020年10月29日)患者出现精神行为异常,表现为夜间穿秋衣、秋裤跑出家门,被派出所发现后通知家人接回。2020年11月3日患者在家中突发意识丧失,伴双上肢屈曲,上下肢伸直抽动,伴双眼上翻、口吐白沫,无舌咬伤、尿失禁,每次持续10余秒钟停止,约半小时后再发作两次。发作停止后患者出现反应迟钝、问话不答,无肢体无力。再次就诊于我科。患者住院后出现躁动、胡言乱语、不配合治疗,予“咪达唑仑间断静脉泵入,奥氮平、奋乃静口服”镇静。2020年11月5日再次复查腰穿一般情况见表2。复查脑电图:轻度异常。11月5日复查头部MRI可见颅内异常信号较前好转(见图4)。再次检测了自身免疫性脑炎相关抗体和脱髓鞘病抗体,结果回报:NMDA抗体 CSF1∶10、血阴性,抗MOG抗体 CSF 1∶10,血1∶100。脑脊液及血AQP-4、抗MBP抗体阴性。临床诊断为自身免疫性脑炎(抗MOG、NDMA抗体阳性),给予免疫球蛋白0.4 g/kg/d 静点共5 d,甲泼尼龙500 mg静点3 d,250 mg静点3 d,120 mg静点3 d,80 mg 静点3 d,后改为泼尼松片60 mg口服,1 w减1片。患者精神症状、认知下降逐渐好转。2020年11月28日神经系统查体:反应略迟钝,余查体未见明显异常。带口服激素出院。

表1 病例1的4次腰穿结果
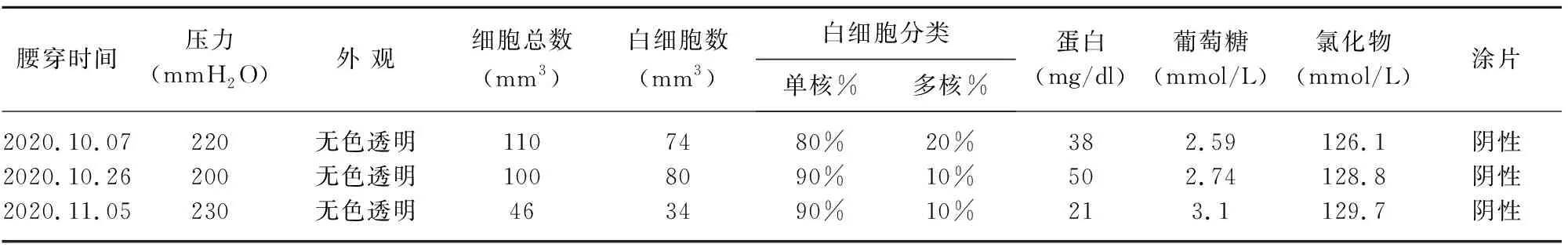
表2 病例2的3次腰穿结果
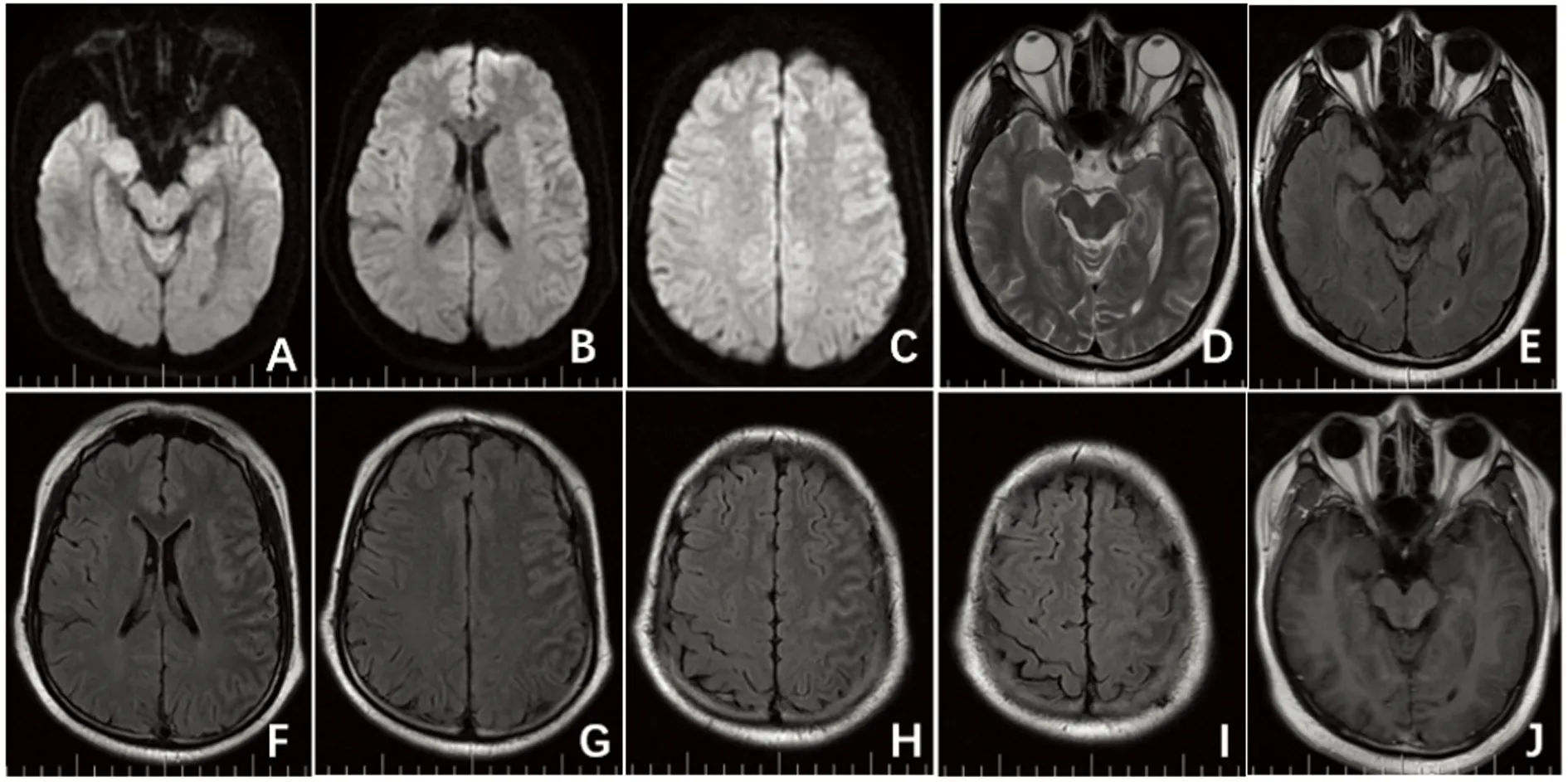
图2 病例2发病2 d(2020年10月6日)头部MRI 可见双侧海马、左侧额颞顶叶皮质异常信号,A-C:DWI呈高信号。D:T2WI呈高信号。E-I:Flair呈高信号。J:MRI增强未见明显强化
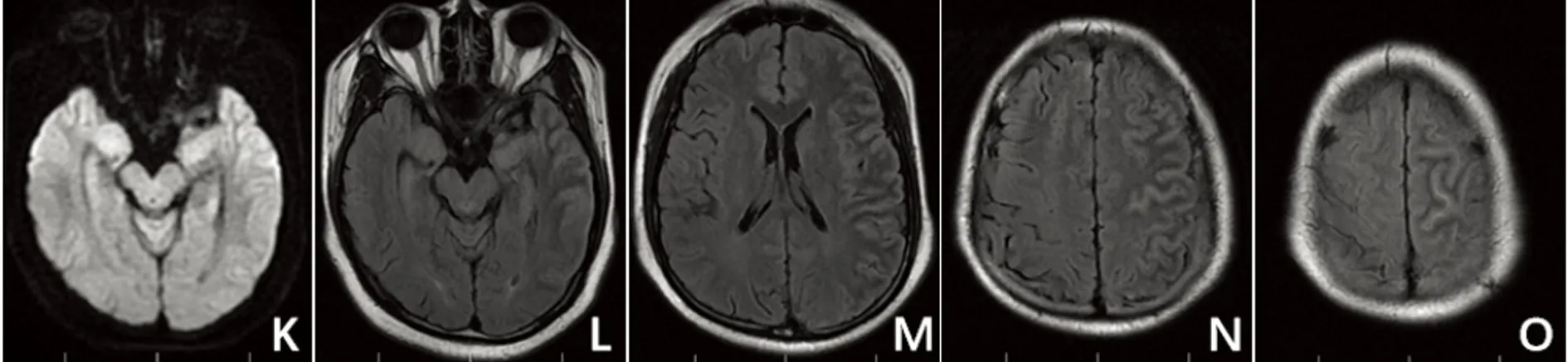
图3 病例2 发病9 d(2020年10月13日)头部MRI 可见双侧颞叶、海马、额叶,左侧顶叶皮质高信号较10月6日增加
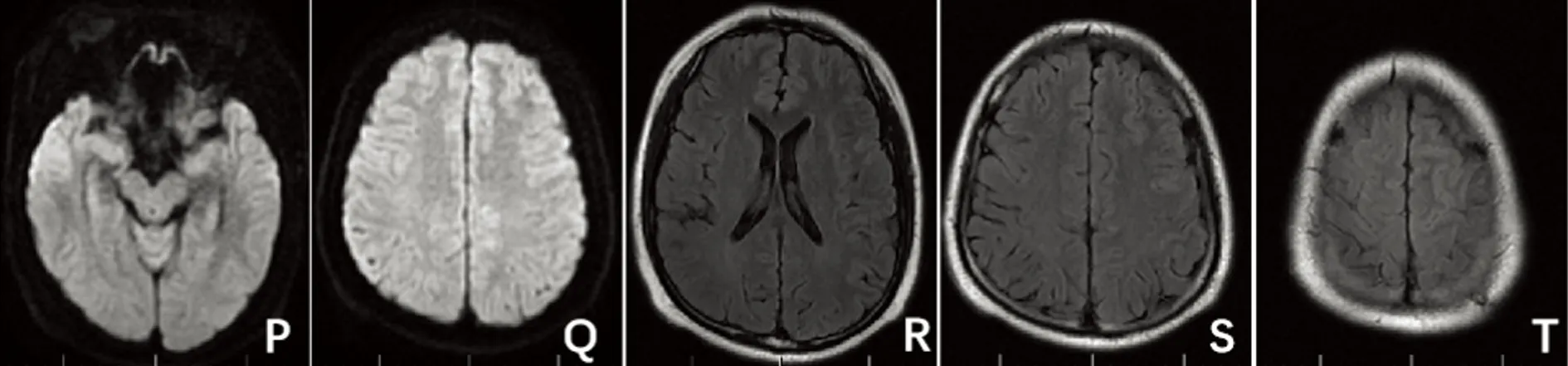
图4 病例2 第2次住院复查头部MRI(2020年11月5日) P-Q:DWI、R-T:Flair均可见双侧皮质异常信号较前好转
2 讨 论
2.1 临床及影像 Du等总结了30例抗NMDA抗体及MOG抗体均阳性的患者,年龄3~48岁,其中女性12例(38%),先出现NMDA抗体阳性47%,MOG抗体先阳性13%,同时阳性40%。临床表现上以行为异常及认知障碍为初始症状最常见(占77%),还可以出现言语障碍(35%)、意识障碍(45%)、视力下降(31%)、肢体瘫痪(42%)、癫痫(32%)、自主神经症状或中枢性低通气(16%)等症状。这30例均未发现肿瘤,影像上幕上病灶占84%,幕下34%,脊髓22%[5]。我们报道的两例患者多次腰穿压力偏高,考虑与患者腰穿过程中未完全放松及患者体型偏胖有关,后期复查腰穿压力正常。两例脑脊液白细胞均明显升高,但<500×106/L,脑脊液蛋白轻度升高,脑脊液葡萄糖、氯化物正常。病例1以发热、头痛起病,数天后出现反应迟钝、智能下降、失语、癫痫发作、右侧肢体偏瘫、中枢性低通气,头部MRI提示双侧额颞顶枕叶广泛大脑皮质异常信号(见图1),检测回报双抗体呈阳性。病例2以“反应迟钝、轻度言语障碍及肢体瘫痪”为主要表现,头部MRI显示双侧额颞顶枕叶皮质异常信号,我们首先想到病毒性脑炎和自身免疫性脑炎,而忽略了MOG抗体的检测,后患者出现癫痫、精神症状,再次检测时出现NMDA及MOG抗体均呈阳性。近年也有报道MOG抗体阳性患者出现单侧或双侧皮质受累,表现为部分性或全面性癫痫发作。我们报道的这两例患者为双侧额颞顶叶枕叶皮质受累,不是MOG抗体阳性患者的典型表现,与但与文献报道的MOG阳性的单侧皮质脑炎影像改变类似[6]。且与文献报道儿童MOG脑炎患者影像改变相似[7,8]。精神症状、癫痫、认知功能减退为脑炎的常见症状,而肢体瘫痪为脱髓鞘病常见症状,当这两种症状同时或先后出现,或影像上存在额颞枕顶叶皮质异常信号时,我们应同时检测这两种疾病的抗体。且当临床怀疑NMDA脑炎时,首次检测呈阴性,经抗病毒治疗后患者再次加重,需复测NMDA抗体,以免漏诊。
2.2 机制 关于自身免疫性脑炎与中枢神经系统脱髓鞘病重叠的机制,目前相关研究较少,已有报道如NMDA与AQP-4,NMDA与MOG均可重叠出现。可能的发病机制有:①少突胶质细胞表面含有NMDAR,胶质细胞与神经元可能产生交叉免疫反应,导致神经元损伤,因此 MOG 抗体更易与NMDAR 脑炎重叠。研究发现,11.9% 的 MOG 抗体阳性患者合并 NMDAR 抗体,明显高于 NMOSD 患者(0.6%)[9]。②病毒性脑炎患者在疾病后期可发生感染后脱髓鞘病,推测可能是病毒感染后启动了针对髓鞘的自身免疫反应。而病毒性脑炎也会继发自身免疫性脑炎,可能与病毒感染后同时启动了针对神经元的自身免疫反应有关[10]。③免疫重建,不同的免疫治疗(包括皮质类固醇治疗)可能会影响免疫状态,在减少剂量或停药时,免疫系统将从免疫抑制中恢复并重建,从而导致免疫细胞攻击自身抗原并导致中枢神经系统炎症反应。该机制在HIV感染患者上多见,但近年发现非HIV感染的免疫缺陷患者在免疫功能恢复期间也会发生[11]。推测这可能与一些应用激素或免疫抑制剂患者检测出多种抗体阳性有一定关系。我们这两例患者病初均有头痛等感冒症状,腰穿脑脊液白细胞数明显升高,且以单核为主,推测病初可能存在病毒感染,诱导了针对髓鞘和神经元的免疫反应,但我们的脑脊液病原学检测未发现特定的病毒,可能与抗病毒治疗一段时间后做检测有关。NMDA与脱髓鞘重叠发生的机制仍有待今后进一步研究。
2.3 治疗及预后 抗NMDA 脑炎治疗分为一线治疗、二线治疗和长程免疫治疗。一线治疗包括糖皮质激素、静脉注射免疫球蛋白和血浆置换。二线药物包括利妥昔单抗与静脉用环磷酰胺,主要在一线治疗效果不佳时应用。长程治疗包括吗替麦考酚酯与硫唑嘌呤,主要用于复发病例,也可用于一线免疫治疗效果不佳的患者和肿瘤阴性的抗NMDA脑炎患者。合并肿瘤的患者,肿瘤切除后病情可获得改善[12]。MOG抗体阳性的脱髓鞘病急性期治疗可应用大剂量激素、免疫球蛋白、血浆置换或免疫吸附等方法。缓解期治疗可口服甲氨蝶呤或硫唑嘌呤,可减低复发率。上述治疗仍不能控制时可考虑利妥昔单抗、奥伐木单抗。那他珠单抗、醋酸格拉默(glatiramer acetate,GLAT)对MOG抗体患者无效,应用β干扰素甚至可能加重病情,故MOG抗体阳性患者不建议使用[13]。双抗体阳性的患者可首选大剂量激素、免疫球蛋白、血浆置换,效果不佳或复发时可以考虑利妥昔单抗、环磷酰胺等。病例1我们应用了抗病毒、免疫球蛋白、大剂量激素冲击等治疗,患者出现中枢性低通气、呼吸衰竭,家属放弃气管插管呼吸机辅助呼吸治疗,最终临床死亡。广泛大脑皮质受累、癫痫持续状态、合并呼吸衰竭提示预后不良。病例2经抗病毒、免疫球蛋白、大剂量激素冲击治疗后,临床症状基本恢复。关于MOG和NMDA重叠患者的治疗及预后缺乏大样本研究,可能是今后研究的重点方向。
猜你喜欢
现代临床医学(2022年1期)2022-02-12
昆明医科大学学报(2021年12期)2021-12-30
天津医科大学学报(2021年4期)2021-08-21
云南医药(2021年3期)2021-07-21
小资CHIC!ELEGANCE(2021年46期)2021-01-11
中国现代神经疾病杂志(2021年6期)2021-01-02
睿士(2020年11期)2020-11-16
VOGUE服饰与美容(2020年9期)2020-09-02
科教新报(2019年26期)2019-09-10
家庭科学·新健康(2019年1期)2019-03-06
