
LAMA3基因突变导致婴儿Herlitz型交界型 大疱性表皮松解症1例
2021-08-27 10:20陈晶晶汪希珂崔玉霞刘征
贵州医药 2021年7期
陈晶晶 汪希珂 崔玉霞 刘征
(1.贵州医科大学,贵州 贵阳 550004;2.贵州省人民医院儿科,贵州 贵阳 550003)
Herlitz型交界型大疱性表皮松解症(Junctional epidermolysis bullosa type Herlitz,JEB-h)是罕见的遗传性皮肤病,为常染色体隐性遗传,通常由LAMA3、LAMB3或LAMC2基因突变引起的[1]。国外报道不多,我国到目前为止仅报道3例[2-4]。该病典型特点是皮肤和黏膜的脆性增加,轻微的摩擦或损伤即可引起水疱或大疱,水疱或大疱破裂后出现表皮糜烂甚至缺损[5]、严重时可累及全身各器官,儿童的发病率约为十万分之二,但死亡率较高[6,7]。平均死亡年龄为5个月,大多数在3岁前死亡[8]。死因多为脓毒症、呼吸衰竭等[9]。本文报道1例Herlitz型交界型大疱性表皮松解症。患儿因“咳嗽、发现全身皮疹1+月”于2020年07月就诊贵州省人民医院儿科。结合患儿的临床资料,以及患儿及父母的全外显子测序的基因检测结果,并复习相关文献,以提高临床医生对该病的认识。报告如下。
1 临床资料
患儿,男,侗族,2月龄,出生1月时全身皮肤出现小水疱,随后水疱增大、融合、破溃、糜烂、结痂,甚至出现皮损。患儿父母非近亲结婚,双方父母及姐姐临床表型均无异常。患儿生后曾患“新生儿脓疱疹、鹅口疮”,饮食无特殊,出生史:出生体重:3600 g,足月顺产,无黄疸、窒息,否认药物过敏史、家族传染病及遗传病史。入院时查体:T:36.5℃,P:142次/分,R:38次/分,SPO2:90%(未上氧下),体重:3.50 kg,神志清楚,精神欠佳,反应欠佳,全身见散在红斑糜烂结痂,臀部枕部头皮见片状红斑、糜烂,少许结痂,少许渗出,未见明显水疱大疱脓疱;口唇欠红润,口腔黏膜见白色凝乳状物附着,不易拭去,咽充血,三凹征阴性,双肺呼吸音粗,双肺闻及少许细湿啰音,心率142次/分,律齐,心音有力,心前区可闻及I-II级杂音。腹软,肝脾未扪及,肠鸣音正常。指趾甲脱落,甲床部分脓疱疹已结痂。神经系统查体无异常。
1.2我院实验室相关检查:血常规:白细胞 31.62×109/L,红细胞 2.91×1012/L,血红蛋白 82.0 g/l,血小板 546.0×109/L,中性粒细胞百分比 65.5%,淋巴细胞百分比 25.0%,中性粒细胞绝对数 20.71×109/L,血沉 50 mm/小时;C反应蛋白(超敏) 196.80 mg/L,抗中性粒细胞胞浆抗体:抗中性粒细胞胞浆抗体(核周型) 1∶10,抗胞浆蛋白酶3抗体 弱阳性 ,抗髓过氧化物酶抗体 阳性(++) ,抗肾小球基底膜抗体 阳性(++),余项阴性;抗核抗体谱:抗着丝点蛋白B抗体 阳性(++)P,抗组蛋白抗体弱阳性P;抗核抗体-胞浆颗粒1∶320;抗核抗体-核颗粒型1∶100 ;抗核抗体-着丝点型1∶100,余项未见明显异常。肝肾功、电解质、心肌酶、甲状腺功能、抗“O”、补体、凝血机制、T-SPOT均未见明显异常。颅脑及腹部CT平扫均未见明显异常。痰培养检出:金黄色葡萄球菌 +++,流感嗜血杆菌 ++++,皮肤分泌物培养检出耐甲氧西林金黄色葡萄球菌,培养发现该菌多重耐药。血培养阴性。患儿全身皮肤出现水疱、破溃、糜烂、结痂、皮损,考虑遗传代谢病可能,故完善全基因组外显子测序(基因测序结果见图1)。经贵州省人民医院伦理委员会批准及家属知情同意,采集患儿及父母静脉血2 mL至于EDTA管中,充分摇匀后备用。应用Qiagen公司血基因组DNA试剂盒(操作按说明书进行),抽外周静脉血基因组DNA进行Sanger测序。采用采用GenCap技术捕获全部编码区外显子及其上下游100 bp邻接内含子区域。运用Illumina HiSeq 2 000测序平台对目标文库进行测序(由北京迈基诺医学检验所完成)。结果显示:患儿LAMA3基因有(c.1591_1602delinsT)纯合突变,导致氨基酸发生移码突变(p.E531fs)。受检父母该位点杂合变异。明确诊断为Herlitz型交界型大疱性表皮松解症,住院期间分别予头孢噻肟钠、哌拉西林他唑巴坦钠、头孢曲松钠、万古霉素抗感染,氟康唑抗真菌,静注人免疫球蛋白调节免疫,甲泼尼龙琥珀酸钠抑制免疫反应,人血白蛋白补充白蛋白、去白细胞悬浮红细胞补充红细胞,重组人牛碱性成纤维细胞生长因子凝胶、复方多粘菌素多软膏外涂疱疹处修复创面以皮肤创面清洁消毒、及保护性隔离等对症治疗后,患儿皮损较前好转,但家属因个人原因拒绝继续住院治疗。
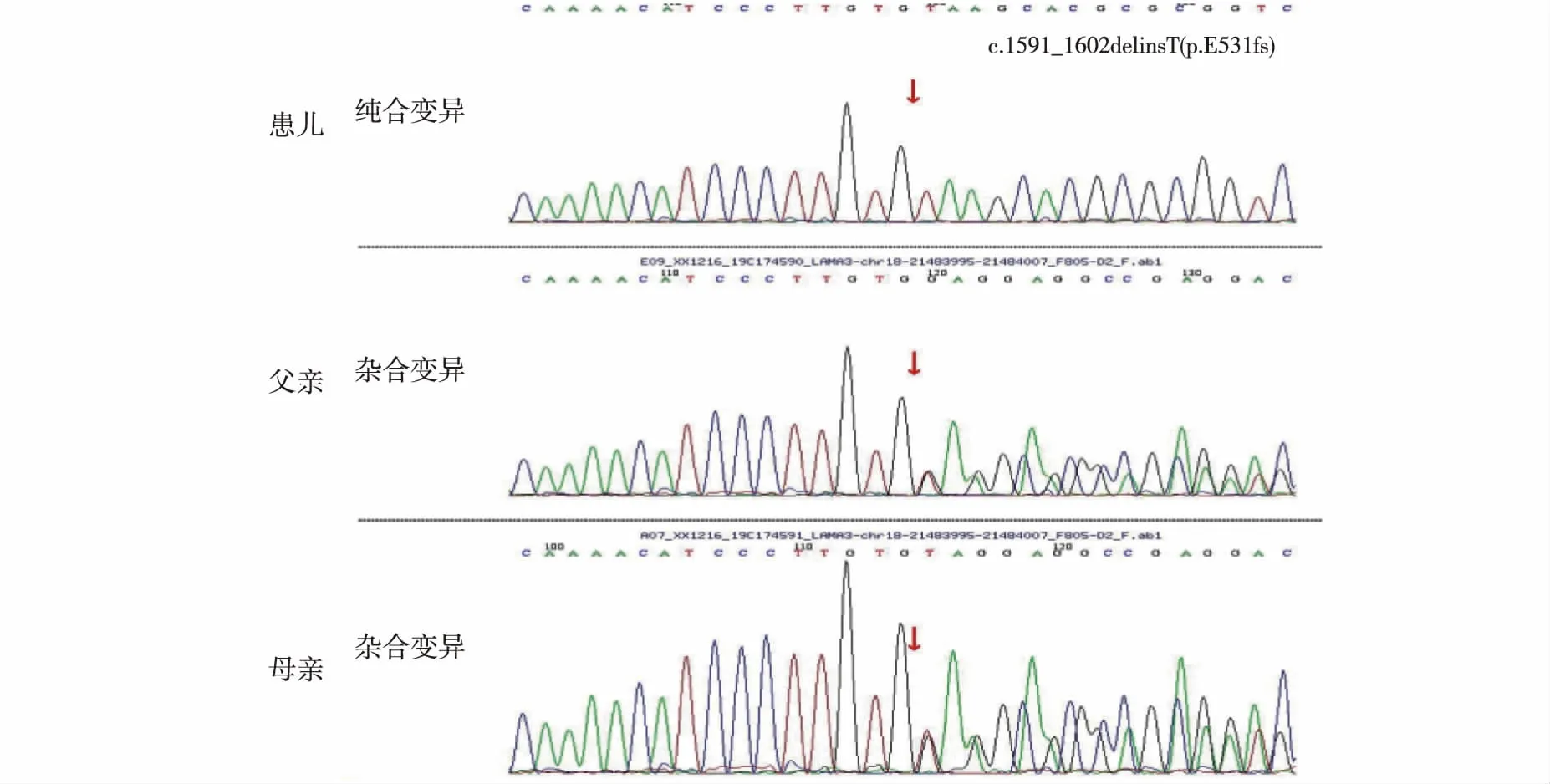
图1 患儿为纯合突变:红色箭头处示1591-602处碱基发生C→T转换;患儿父母红色箭头处示杂合突变
2 基因检测结果
患儿LAMA3基因编码区第1591号碱基与1602号碱基之间发生了缺失,导致氨基酸发生移码(p.E531fs),该位点突变在国内为首次报道,该变异是纯合变异,来源于杂合携带父母,正常人中频率为-,根据ACMG指南对致病性进行评判,判断为Likely pathogenic;PVS1+PM2(PVS1: 该变异为零效变异(移码突变),可能导致基因功能丧失;PM2:在正常人群数据库中的频率为-,为低频变异)。
3 讨 论
大疱性表皮松解症 (epidermolysis bullosa,EB)是一种罕见的常染色体显性或隐形遗传的皮肤病,基本病理改变是角质形成细胞或皮肤基底膜蛋白缺失或结构异常。临床主要有单纯型大疱性表皮松解症(epidermolysis bullosa simplex,EBS) 、营养不良型大疱性表皮松解症(epidermolysis bullosa dystrophica,DEB)、交界型大疱性表皮松解症( epidermolysis bullosa junctional,JEB)和Kindler综合征几种类型[10,11]。JEB-h属于重型JEB,又称为全身性JEB[12],患儿多在出生时可出现广泛的皮肤黏膜水疱[1]。但程度较其他类型更严重,预后更差,致死率也更高[13]。
本例患儿为2+月小婴儿,出生1个月时全身皮肤出现小水疱,随后水疱逐渐增大、融合、破溃、糜烂、结痂,甚至出现皮损。全基因组外显子测序结果提示患儿LAMA3基因有1个纯合突变(c.1591_1602delinsT纯合突变),导致氨基酸发生移码突变(p.E531fs)。患儿父母该位点杂合变异。根据患儿临床资料及全基因组外显子测序结果,可明确诊断为Herlitz型交界型大疱性表皮松解症。目前国内上海[4]及青岛地区[3]有关于JEB-h的报道,但发病年龄均大于本例,并且基因突变类型也与本例不同。目前尚无与本例基因突变一致的相关报道。对于基因缺陷病,产前诊断只能够干预患儿的出生,不能用于已出生患儿的诊断[7]。而传统的 Sanger 测序耗时、费用高,临床中的应用有限[14]。新一代测序技术促进了全基因组外显子测序的发展。全基因组外显子测序的原理是利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法,该方法灵敏度高,只需对1%基因组进行测序就能发现外显子区绝大部分疾病相关变异。主要有目标区域序列的富集、DNA 测序、生物信息学统计3步[15]。在基因病的研究和临床疾病的诊断中应用广泛[16]。成为诊断基因病的“新标准”[17]。针对EB的治疗,临床上多为对症治疗及局部皮肤护理来促进部分伤口愈合,一定程度的缓解临床症状[20],目前尚无特效药物。国内有报道使用糖皮质激素可以缓解皮肤疱疹[21],本例患儿联合使用糖皮质激素抑制免疫炎症反应治疗后皮损也较前有所好转。
综上述,本文报道1例LAMA3基因突变导致婴儿Herlitz型交界型大疱性表皮松解症,为LAMA3基因(c.1591_1602delinsT)纯合突变,导致氨基酸发生移码突变(p.E531fs)。患儿父母该位点杂合变异。对于临床表型疑似病例进行全基因组外显子测序可明确诊断该病,但目前仍无特效药物用于该病的治疗。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
临床骨科杂志(2022年3期)2022-06-24
建材发展导向(2021年14期)2021-08-23
中国生殖健康(2020年4期)2021-01-18
爱你·健康读本(2019年8期)2019-11-22
祝您健康·文摘版(2019年4期)2019-06-11
中国生殖健康(2018年4期)2018-11-06
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
中国房地产业(2016年9期)2016-03-01
湖北农业科学(2014年11期)2014-09-10
