
A TP敏感性钾通道的研究进展
2021-08-19 01:03:52邵丽静赵伟综述陈渊审校
中国生物制品学杂志 2021年8期
邵丽静,赵伟 综述,陈渊 审校
浙江农林大学林业与生物技术中药学科,浙江 杭州 311300
1983年,NOMA[1]利用膜片钳技术,首先发现了ATP敏感性钾通道(ATP-sensitive potassium channel,KATP),这类通道会随着细胞内ATP浓度的升高而活性降低。随后人们在胰岛β细胞、血管平滑肌、线粒体内膜等组织中也发现该通道。根据细胞不同部位的分布,KATP通道可分为位于细胞膜上的KATP通道和位于线粒体膜上的KATP通道。KATP通道分布于人体多个器官,其结构等变化影响着人体的生理功能。研究发现,KATP通道在改善心脏缺血预适应,调节微血管舒缩状态、控制胰岛素分泌等多种生命活动中发挥着重要作用。本文现对KATP的调控方式、分子结构及生理功能等方面的研究进展作一综述。
1 K ATP通道调控
KATP通道是内向整流钾离子通道分类中的一种。首先,KATP通道是弱的内向整流钾离子选择性通道(weak rectification),通道活动的显著特征是呈簇状开放,其主要调控特征为胞质ATP快速可逆关闭,以及核苷酸三磷酸酯和二磷酸酯激活;其次,通道可被甲苯磺丁脲(tolbutamide)等磺脲类化合物所抑制,也可被KATP激动剂(KATPchannel openers),如二氮嗪(diazoxide)等激活。
KATP通道的标志是被ATP强烈抑制,因此ATP和ADP为通道的主要门控分子。胞内ATP对KATP通道具有双向调节的作用,即当细胞内ATP浓度增高时,KATP通道活性被抑制,当ATP浓度降低时,KATP通道又被激活。而在通道关闭状态下,ATP在Mg2+的协助下,可使通道恢复开放。在没有Mg2+的情况下,核苷酸与Kir6亚基直接作用来抑制KATP通道活性[2]。在Mg2+存在下,通过与SUR亚基的相互作用[3],ATP和ADP均会刺激通道活性[4]。另外,膜磷脂,特别是磷脂酰肌醇二磷酸[phosphatidylinositol(4,5)bisphosphate,PIP2]通过结合Kir6.2亚基,调节KATP活性[5],而PI-PLC(磷脂酶C)通过水解PIP2降低KATP通道活性。
ATP的结合位点至今仍有争议。早在1995年,AGUILAR等[6]认为,含有2个核苷酸结合域(nucleotide binding domain,NBD)的SUR是ATP结合的主要抑制位点。然而在1997年,TUCKER等[7]发现,Kir6.2截断26个C-末端残基(保留信号),仍可在细胞质膜中形成功能四聚体通道,即使在无SUR的情况下钾离子也可以通过细胞膜,并且与野生型通道一样,ATP同样可以强烈地抑制该通道,表明ATP抑制是由与Kir6.2结合引起的。由于Kir6.2中的ATP结合位点与经典的核苷酸结合位点(如SUR的NBD)不同,ATP不能被Kir6.2水解,表明通道门控与ATP水解无关。ARAKEL等[8]研究证明,SUR1是ADP传感器,由于共有位点对ADP的亲和力可能较ATP高,因此,即使在生理ATP浓度设定下,该通道也可能潜在地感知ADP水平的微小变化。虽然ATP抑制是Kir6.2固有的,但SUR1对ATP抑制作用产生两种相反的调节:SUR1的存在增强了ATP抑制的效力;SUR1的存在可使MgADP解脱ATP抑制。
此外,长链酰基辅酶A分子(LC-CoA),脂肪酸β氧化的中间产物,也被证明以与膜磷脂酰肌醇(phosphatidylinositol,PI)类似的方式调节通道活性。介导PIP2活化的Kir6.2上的相同残基也有助于LC-CoA的刺激作用,提示Kir6.2是LC-CoA作用的主要位点。激动剂依赖性蛋白激酶A(protein kinase A,PKA)磷酸化可控制平滑肌和胰腺KATP通道的开放:Kir6.2亚单位有2个一致的PKA磷酸化位点,当磷酸化发生,增加了通道开放概率时,在人类SUR1中,一个独特的组成型磷酸化PKA位点,可用于增加通道的表达并降低通道开放概率。蛋白激酶C(protein kinase C,PKC)对自然心室KATP通道具有混合作用,通过磷酸化Kir6.2亚单位中高度保守的T180残基[8],在低ATP浓度下具有抑制作用,但在高ATP浓度下可激活KATP通道。Kir6.1/SUR2B通道也同样由于Kir6.1残基的磷酸化而被急性PKC抑制,但SHI等[9]的实验发现,Kir6.2/SUR2B通道活性不受PKC影响,因此PKC的具体作用尚需进一步研究,但可以肯定的是,PKA包括环磷酸腺苷(cyclic adenosine monophosphate,cAMP)和Epac2等,几乎普遍被接受是GLP-1(glucagon-like peptide-1)刺激的胰岛素分泌的主要机制[10]。
PIP2、ATP和ADP是细胞内3种天然活性分子,互相作用调节KATP通道的活性,PIP2是活性所必需的,ATP抑制活性,ADP增强活性。AZIZ等[11]在实验中发现,ATP似乎负向调节PIP2,并且ADP可克服ATP抑制,同时,还发现了ATP结合位点和PIP2结合位点与SUR(是ADP结合位点)的结构互联性,这似乎与它们的功能依赖性相关。
2 K ATP特征及分子结构
内向整流钾通道是一大类非电压门控K+通道。具有两个基本功能:稳定的静息电位并促进钾的膜转运。所有Kir通道均是由4个相同或相似的亚基组成的四聚体,形成同源或异聚体通道。天然的Kir1.X、Kir2.X和Kir6.X可能以同种四聚体的形式存在于体内,而Kir3.X通道主要产生异四聚体,由大脑中的Kir3.2和Kir3.4亚基以及心脏中的Kir3.1和Kir3.4亚基形成。
自从1993年KUBO等[12]通过表达克隆分离出第1个Kir通道基因以来,目前发现至少有17个真核Kir通道基因分布在7个亚科(Kir1~7)和多个原核亚型(Kirbac1.1~9)中;而Kir通道按功能通常可分为4类:经典Kir通道(Kir2.X)、G蛋白门控Kir通道(Kir3.X)、ATP敏感钾离子通道(Kir6.X)和K+传输通道(Kir1.X、Kir4.X、Kir5.X和Kir7.X)。Kir通道系统发生树见图1[13]。Kir通道分布广泛,已经在许多细胞类型中被发现,并且具有变化的整流特性。由于这些通道的整流特性,在膜电位正电位约-40 mV时,仅使得极少量的电流流过细胞膜;而小于-40 mV时,使细胞处于高电导状态,允许细胞膜电位在静息电位附近保持稳定。这一特性可以使细胞在正电压下大大降低电导,避免动作电位的短路。

图1 Kir通道的系统发生树及分类Fig.1 Phylogenetic tree and classification of Kir channel
1998年,DOYLE等[14]研究发现了细菌KCSA通道的结晶,揭示了离子通道家族成员(包括Kir通道)共有的基本架构支架。KCSA是由2个跨膜螺旋,经细胞外环桥接,形成狭窄的孔并具有离子选择性。KCSA虽然是仅有2个α螺旋的细菌钾离子通道,但也正是这个晶体结构的解析,首次揭示了离子通道的选择性和离子跨膜通透的基本结构,同时也证明了以前的理论推测。
2005年,KUO等[15]通过电子低温显微镜解析全长原核生物Kir通道同源物KirBac1.1和Kir-Bac3.1的结构,揭示了一个扩展的细胞质孔和配体结合位点的可能所在的位置,以及真核生物Kir通道的一些独特性质。这一结果与数十年来的预测结果相符,为后来的研究者奠定了坚实的结构基础。也正是基于这一研究结果,才有了NISHIDA等[16]的Kir3.1及Kir2.1的连接N-和C-末端结构域的结晶。
随后,人们发现4类Kir通道均具有相同的1个基本结构:—NH2和—COOH末端的两个跨膜螺旋(M1和M2)以及由大约30个氨基酸的短链连接形成的细胞外成孔区(H5)。Kir6.2是其中的一个典型,其具有2个跨膜螺旋(M1和M2),由一个凹入的孔螺旋和环分开,后者包含一个K+通道特征序列(TVGYG),赋予了通道离子选择性[17],图2显示了K+结合位点的线性阵列。TVGYG以球棒表示法表示。在过滤器的细胞外和内部末端,水分子围绕K+。当离子进入过滤器时,它们的水合壳逐渐被与选择性过滤器(位置0~4)的骨架羰基的相互作用取代。过滤器在位置1和3或位置2和4同时包含2个离子。

图2 KcsA选择性过滤器显示的K+结合位点的线性阵列Fig.2 KcsA selectivity filter showing linear array of K+binding sites
最近,一种先前未鉴定的跨膜蛋白175(TMEM-175)被鉴定为是真正的溶酶体钾离子通道,其功能对于设定溶酶体膜电位和维持pH稳定非常重要。TMEM175与经典四聚体K+通道无序列同源性,缺少TVGYG选择性过滤器,同时,LEE等[18]还采用了非传统的方式确定了CmTMEM175(TMEM175 from Chamaesiphon minutus,微型管孢藻的TMEM175)的结构并精确至3.3Å,同时为K+通道家族关于选择性方面的研究又丰富了内容。另外,TMEM175的缺乏还被证明在帕金森病的发病机制中起关键作用。
SUR属于ABC转运蛋白(ATP-binding cassette transporter,ABC)家族,但缺乏内在的转运活性,是KATP通道的重要调节亚基,通过组织分布和mRNA表达可分类为SUR1、SUR2A和SUR2B 3种亚型。SURX有17个跨膜螺旋(M),由3个部分组成:5个螺旋构成的跨膜区(transmembrane domain,TMD)和TMD0(M1-M5)、TMD0旁边的L0环以及TMD1(M6-M11)和TMD2(M12-M17)组成。与其他ABC蛋白一样,SUR1的ABC核心是域交换的,因此每个半转运蛋白由来自TMD1和TMD2的螺旋组成。SUR1的核苷酸结合位点(nucleotide binding site,NBS)是不对称的,其具有一个催化(ATP水解)能力的NBS(NBS2)和一个不能催化ATP水解的位点(NBS1)。TMD0可直接与Kir6.2联系,TMD0/L0和Kir6.2之间相互作用位点的定位有助于解释这些残基在与孔亚单位共存时如何促进Po的增加。TMD0与TMD1之间的2个核苷酸结合折叠(NBF)与水解的ATP结合,并且可能由于细胞内代谢变化而起调节作用[19]。与Kir组不同,SUR组可用于与MgADP组合打开通道。SUR1是抑制KATP促进胰岛素分泌的磺脲类药物的靶标。
位点定向诱变和详细的电生理学已经确定了Kir6.2上抑制性ATP结合位点的大致位置在PIP2[20]上,可通过与Kir3.2(GIRK2)的结构和序列比对来鉴定Kir6.2中推定的PIP2结合残基位置。KIN等[21]研究的新结构揭示了抑制位点相对于假定的PIP2结合位点(KPIP)的精确位置,并且观察到了ATP结合位点、PIP2结合位点与SUR的结构之间的相互关联性,并提出这些关联性均与它们的功能依赖性相关。
KATP复合物大多还只有低分辨率的结构信息,包括连续的SUR1-Kir6.2的负染色EM结构(18Å)、四聚体SUR2B的EM结构(21Å)以及NBD的小角度X射线散射和核磁共振研究[22]。但目前,LI等[23]使用冷冻电子显微镜解析了胰岛β细胞KATP通道的中等分辨率(5.6Å)结构,以及KIN等[21]在存在Mg2+和核苷酸的情况下所解析的结构(螺旋桨形状确定为5.6Å分辨率,四叶形状确定为3.9Å分辨率),均更进一步地揭示了KATP的结构模式。
3 K ATP通道的药理学和病理学
由于KATP通道能够通过感知细胞内的ADP和ATP比值来开放和闭合通道,静止时,KATP激活引起膜超极化,而其抑制作用则产生膜去极化,因此,可通过将细胞代谢与质膜的电活动相联系来研究KATP通道。历年来,人们广泛地进行了KATP通道在葡萄糖稳态和缺血中保护作用的研究,发现了磺酰脲类药物可以降低血糖。随后人们还发现了通道的一些其他作用,如通过KATP通道可保护中风后神经细胞的凋亡[24]、人类记忆也与大脑中的KATP通道有关[25]、KATP通道还可调节男性生殖行为[26]等。
由于KATP通道是一种广泛存在于胰腺、线粒体等部位的钾离子通道,因此,利用KATP通道的激动剂或者拮抗剂调节通道的开放及闭合,已经成为该类组织相关疾病的一种治疗方法。如经典的KATP通道激动剂,如二氮嗪和吡那地尔,已被用于治疗婴儿高血压、心绞痛和高胰岛素症,而拮抗剂,如磺酰脲类已被确定为抗糖尿病药物。激动剂二氮嗪除了可以打开KATP通道外,还可抑制琥珀酸脱氢酶(succinate dehydrogenase,SDH),并且有研究发现,这种酶还是Krebs循环中的关键[27]。
3.1 KATP药理学 根据KATP通道的特性,想要闭合或者打开通道,首先可以选用针对性的药物阻断或激活;其次可通过某些物质来调节激动剂或拮抗剂对通道的活性,从而改变特定通道的开放概率。如PIP2、PI-PLC、PH和LC-CoA等均可调节通道的活性,此外还可通过改变通道对ATP的敏感度从而调节通道活性,但KATP通道对ATP的敏感性没有一个特定的参数范围,可能会随着膜组成的变化而变化。
SUR亚基决定了通道对多种激动剂和拮抗剂的敏感性。如格列本脲可抑制所有SUR亚单位,但抑制程度取决于细胞内核苷酸浓度,因此也就取决于细胞的代谢状态。磺酰脲类也可与Kir6.2亚基相互作用,但亲和力较低,不如对SUR亚单位的作用[28]。也正是由于这种对不同亚基亲和力的差异性,使得磺酰脲类药物可用于靶向特定的KATP通道,如微摩尔浓度下的甲苯磺丁脲仅靶向SUR1通道,而格列本脲和美格利汀则可阻断SUR1和SU2A通道[29]。
同样,不同激动剂也可能作用于不同SUR亚单位,这种特异性也是一种重要的药物研究方向。针对不同的SUR亚型,KCOs对KATP通道也具有不同的亲和力。大多数KCOs对SUR2A和SUR2B亚型具有较高的亲和力,但MOREAU[30]的研究发现,二氮嗪主要影响SUR1亚型。该研究还表明,2个位于SUR2A受体螺旋17中的残基L1249和T1253与非二氮嗪类的激动剂的结合有关。并且MOREAU等[31]的另外一项研究表明,当这些残基发生突变时,SUR2A也就失去了被激动剂调节的能力。此外,UHDE等[32]的研究表明了连接螺旋13~14的环区域对通道调节的重要性。以上两项研究强调了TMD2结构域在KATP通道激动剂与SUR2亚型选择性结合中的重要作用。
KCOs的活性与MgATP和MgADP的存在也有关。如在MgADP存在下,对SUR1有特异作用的二氮嗪可与SUR2结合。此外,已经观察到KCOs结合可通过NBD被MgATP正向调节[33]。另外这些激动剂均需要可水解的ATP才能发挥作用,这也说明激动剂在核苷酸结合折叠处起到稳定或增强ATP水解的作用。
3.2 KATP病理学
3.2.1 心肌细胞KATP通道 在正常稳定的代谢条件下,心肌细胞内的ATP浓度足以让肌膜KATP通道几乎完全关闭,也就是几乎不引起细胞兴奋。当心肌细胞中的KATP通道处于某种应激状态,如缺氧、代谢抑制时,通道会开放来保护细胞免受Ca2+过载的损伤,KATP电流激活导致动作电位缩短,减少Ca2+进入并抑制收缩性,从而减少VSM(vascular smooth muscle)的能量消耗并加速缺血后的恢复[34]。
Kir6.1是血管平滑肌KATP通道的基础,在血管反应性和血压控制中具有关键作用[35]。研究发现,Kir6.2基因敲除小鼠不能进行剧烈运动,并在高血压,压力超负荷或缺血的情况时表现出心脏重塑、心力衰竭和死亡的现象。OLSON等[36]研究发现,房颤和心力衰竭与SUR2A功能缺失突变有关,Kir6.1敲除和SUR2敲除的小鼠会出现人类变异型心绞痛的许多临床特征,包括基线高血压、冠状动脉血管痉挛和突发性心脏死亡。BARAJAS-MARTÍNEZ等[37]研究发现,在一些早期复极综合征和Brugada综合征患者中,Kir6.1(S422L)会有一个功能突变,但VEERAMAH等[38]的研究表明,S422L可能是一种良性变体。在婴儿猝死综合征的病例中,Kir6.1的突变会导致KATP通道活性降低[39]。SUR2基因的突变还与Cantù综合征(Cantùsyndrome,CS)有关。
3.2.2 胰腺KATP与心脏KATP通道不同,由于SUR1和SUR2A赋予的增强刺激作用,胰腺KATP通道由Kir6.2和SUR1组成[40],见图3,Kir6.2通道的4个亚基与磺酰脲受体的4个亚基缔合,形成功能性KATP通道。Kir6.2具有2个跨膜螺旋(TM1和TM2)和1个包含ATP结合位点的大胞质域。SUR亚基由3个跨膜螺旋(TMD0、TMD1和TMD2)和2个核苷酸结合结构域(NBD1和NBD2)组成,其中包含Walker A、Walker B和Linker L共有序列。通过调节膜的兴奋性将血糖水平与胰岛素分泌相关联。同时,PIP2影响胰岛β细胞KATP通道,不仅通过其对Kir6.2的作用,还通过隔离Syn-1A(syntaxin-1A)来调节Syn-1A的可用性及其与PM(plasma membrane)上SUR1的相互作用[41]。由于SUR1刺激作用增强以及在禁食期间胰岛β细胞中相对较低的ATP与ADP比率,胰腺KATP通道形成负膜电位。但在餐后血糖水平升高、葡萄糖被吸收代谢之后,ATP与ADP比率就会增加,KATP通道的活性降低从而通道关闭,导致细胞膜去极化以及电压门控钙离子通道的激活,随后胰岛素分泌,从而达到降血糖的效果。由于这些通道在葡萄糖刺激胰岛素分泌中起关键作用,因此对Kir6.2或SUR1的基因修饰可能导致胰岛素分泌紊乱。
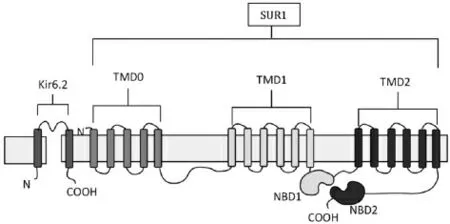
图3 KATP通道的示意图Fig.3 Schematic illustration of KATP channel
KATP通道的突变可导致胰岛功能失效,这些突变可分为2类:导致通道表达减少的突变和降低通道开放概率的突变。实际上,任何NBS的突变均可能导致胰岛素分泌疾病,包括婴儿期持续性高胰岛素高血糖症或新生儿糖尿病[42]。
先天性高胰岛素血症(congenital hyperinsulinism,CHI)为一种复杂病症,是由胰岛β细胞胰岛素分泌不受调节而导致严重低血糖导致的。CHI的潜在机制包括质膜中KATP通道的完全丧失或MgADP对KATP通道激活的损害,后者可调节KATP通道的闭合状态,导致永久性膜去极化和胰岛素分泌。KATP通道的ABCC8/KCNJ11基因突变可导致最严重的CHI。研究发现,几种异常变体在调节胰岛素释放方面具有特别重要作用,可能导致症状较轻的高胰岛素血症低血糖[43]。另一方面,据报道,Kir6.2残基23(E23K)中遗传变异的存在在2型糖尿病患者中很常见(E23K导致糖尿病发展的确切机制尚不清楚)[44]。在2000年,KOSTER等[45]就发现,KATP通道的功能获得突变在小鼠模型中引起葡萄糖耐受不良。该研究表明,KATP通道中的功能获得性突变可能降低胰岛素分泌,导致人类糖尿病的发展。
自2004年报道了第1例KATP通道中出现功能获得性突变的新生儿糖尿病以来,新生儿糖尿病患儿数目逐年增加,患病率估计为1/250 000,其特征在于出生后6个月内会发生糖尿病[46]。近一半的新生儿糖尿病病例是由Kir6.2(KCNJ11)和SUR1(ABCC8)突变引起的,其中约31%的病例是由KCNJ11突变引起的,13%是由ABCC8突变引起的[47]。这些患儿中仅有约20%的患儿具有KATP通道突变患者出现的神经症状。
新生儿糖尿病根据症状的严重程度分为以下4种亚型:短暂性高血糖症[短暂性新生儿糖尿病,(transient neonatal diabetes mellitus,TNDM)];永久性高血糖症[永久性新生儿糖尿病(permanent neonatal diabetes,PNDM)];DEND综合征(neonatal diabete syndrome),存在严重的神经系统症状,如发育迟缓、癫痫和肌肉无力;DEND综合征症状无癫痫[中间型DEND综合征(intermediate neonatal diabetes syndrome,iDEND)]。
胰岛β细胞中的KATP通道亚基中的功能丧失突变是CHI的基础,DEND综合征会以最严重的永久性疾病形式延伸至胰腺以外的神经元或其他组织,使患者出现运动和智力发育迟缓及癫痫和新生儿糖尿病。DEND的胰腺外症状归因于肌肉、神经和脑中的KATP过度活动,这也突出了Kir6.2和SUR1在胰腺外组织中的作用。一些患有DEND综合征致病突变的患者表现出注意力缺陷多动障碍(attention-deficit hyperactivity disorder,ADHD)、自闭症或睡眠障碍的症状[48]。在小鼠模型中,具有导致DEND的V59M突变的神经组织的小鼠出现的多动、探索性行为增加和焦虑减少,与ADHD和孤独症的症状均一致[49]。但这些精神疾病发展的确切机制仍不清楚。
报道的Kir6.2突变主要是显性杂合子,但也有少数为纯合突变,而SUR1中的突变既可以是显性遗传也可以是隐性遗传[50]。新生儿糖尿病引起的Kir6.2突变的功能研究表明,这些突变降低了ATP抑制KATP通道的能力[51]。由于ATP敏感性的微小变化而发生的KATP通道活性的这些微小变化可改变β细胞电活动和胰岛素分泌,从而导致患者糖尿病。这种降低的ATP敏感性的分子机制取决于Kir6.2突变的位置。
3.2.3 中枢神经系统中的KATP通道 KATP通道在大脑的不同区域均有表达,如GABA能神经元主要表达SUR1和Kir6.2亚型,多巴胺能神经元表达SUR1/Kir6.2和SUR2B/Kir6.2复合物。此外,在下丘脑中也发现了SUR1和kir6.1亚型。
中枢神经系统的损伤通常会伴有一定的代谢紊乱、细胞凋亡、脑组织水肿或者坏死等症状。在中枢神经系统损伤的过程中,KATP通道的开放对某一类疾病可起到缓解病情进展的作用。通过使KATP通道开放不仅可降低神经元的兴奋性,还可减少兴奋性氨基酸的释放从而减少能量代谢,同时减少了线粒体及其他细胞器的损伤,并且通过阻断细胞凋亡通路来减少缺血缺氧时神经细胞凋亡情况的发生。但KATP通道的开放同样会加速病情的进展,如在脑卒中的早期,通道的开放会影响小胶质细胞的神经保护作用,进而导致中枢神经系统更为严重的损伤。
YAMADA[52]研究了KATP通道在脑组织中的作用,通过比较低氧对野生型和基因敲除型小鼠脑组织的影响发现,在缺氧(缺氧所致)情况下,野生型小鼠的神经元活性降低,而对于Kir6.2基因敲除的小鼠,活性增加。野生型小鼠的实验结果表明,在代谢应激条件下,由于ATP浓度的降低,KATP通道开始变得更加活跃,对缺血具有保护作用。
ESMAEILI等[53]研究发现,Aβ(β-淀粉样蛋白)的施用可在啮齿动物中改善抑郁症和焦虑症状,表明KATP通道的拮抗剂可能是治疗阿尔茨海默病(Alzheimer disease,AD)患者情感障碍的合理治疗策略。此外,在AD模型发生记忆缺失的动物中,使用低于临床剂量的KATP通道的激动剂及拮抗剂,可不同程度地限制Aβ诱导的记忆缺失以及海马网络长时程突触可塑性不平衡的表现。SALGADO-PUGA等[54]研究发现,甲糖宁对于抵抗Aβ诱导的记忆缺失的效果比二氮嗪好。并且,还有研究发现,KATP通道在帕金森病(Parkinson disease,PD)中同样也发挥着重要作用[55],但其具体的作用机制尚不明确。
4 小结与展望
KATP通道广泛分布于人体内,其可参与人体中的一系列生理、生化反应。如KATP拮抗剂可通过关闭胰腺β细胞中的KATP通道,刺激胰岛素分泌来调节血糖。KATP激动剂作用靶点多,其中的许多靶点均具有一定的临床研究价值,如对PD、AD和CHI等。但目前无论是KATP拮抗剂还是KATP激动剂所面临的主要问题是其对人体组织的选择特异性较差,因此开发具有高选择性的药物将会是未来的研究方向。
猜你喜欢
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
世界最新医学信息文摘(2020年68期)2020-12-25 11:55:27
生物学通报(2019年3期)2019-02-17 18:03:58
中成药(2018年10期)2018-10-26 03:41:22
天然产物研究与开发(2018年6期)2018-07-09 06:01:46
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:13
医学研究杂志(2015年5期)2015-06-10 06:43:26
赤峰学院学报·自然科学版(2013年4期)2013-07-31 22:01:40
中国医学科学院学报(2013年6期)2013-03-11 20:26:01
