
人干扰素α2a注射液中对羟基苯甲酸甲酯和对羟基苯甲酸丙酯含量反相高效液相色谱检测方法的建立及验证
2021-08-19 01:03何珊于雷贾丹靳征袁杰史新昌裴德宁
中国生物制品学杂志 2021年8期
何珊,于雷,贾丹,靳征,袁杰,史新昌,裴德宁
1.沈阳三生制药有限责任公司,辽宁 沈阳 100271;2.中国食品药品检定研究院,北京 100050
人干扰素α2a注射液是由高效表达人干扰素α2a基因的大肠埃希菌经发酵、分离和高度纯化后获得的人干扰素α2a原液制成,辅料包括吐温80、枸橼酸钠、枸橼酸、对羟基苯甲酸甲酯、对羟基苯甲酸丙酯、乙二胺四乙酸二钠等,临床适应症为淋巴或造血系统肿瘤和某些病毒性疾病,已收录于《中国药典》三部(2020版)。
对羟基苯甲酸甲酯和对羟基苯甲酸丙酯为无色结晶或白色结晶性粉末,主要用作食品、化妆品、医药的杀菌防腐剂,可破坏微生物的细胞膜,使细胞内的蛋白质变性,并可抑制微生物细胞的呼吸酶系及电子传递酶系的活性,从而起到杀菌防腐的作用[1-3]。在人干扰素α2a注射液的生产中,常联合使用对羟基苯甲酸甲酯和对羟基苯甲酸丙酯,以提高其防腐效能[4-5]。药品中防腐剂含量过低起不到抑菌效果,过高则会对人体产生毒害作用,因此应严格控制其含量。根据《中国药典》三部(2020版)《重组人干扰素α2a注射液》项下规定:如制剂中含有对羟基苯甲酸甲酯,其含量应为0.04%~0.10%,如制剂中含有对羟基苯甲酸丙酯,其含量应为0.004%~0.010%,测定方法为气相色谱法(通则3116)[6]。检定过程中,发现该方法存在精密度低、检测时间长等缺点[7],为克服这些缺点,本研究建立了测定人干扰素α2a注射液中对羟基苯甲酸甲酯和对羟基苯甲酸丙酯含量的反相高效液相色谱(reversed-phase high performance liquid chromatography,RP-HPLC)法,并对其进行验证[8-9]。
1 材料与方法
1.1 主要试剂及仪器 重组人干扰素α2a注射液(批号:201804026、201807032、201904040、2019040-41、201904042和201904044)由沈阳三生制药有限责任公司提供;对羟基苯甲酸甲酯对照品(批号:100278-201906)、对羟基苯甲酸丙酯对照品(批号:100444-201804)购自中国食品药品检定研究院;不含对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的重组人干扰素α2a注射液空白制剂由沈阳三生制药有限责任公司质量检验部实验室制备;冰乙酸(分析纯)购自沈阳化学试剂厂;甲醇(色谱纯)购自美国SIGMAAldrich公司;e2695高效液相色谱仪、e2489紫外检测器购自美国Waters公司;Phenomenex C18色谱柱(5μm,250 mm×4.6 mm)购自美国Phenomenex公司;Grace C18色谱柱(5μm,250 mm×4.6 mm)购自美国Grace公司;Agilent C18色谱柱(5μm,250 mm×4.6 mm)、HP-1色谱柱(30 m×0.530 mm×2.65μm)、7890B气相色谱仪购自美国Agilent公司。
1.2 R P-H PL C法的建立
1.2.1 色谱条件 色谱柱:Phenomenex C18柱(5μm,250 mm×4.6 mm),流动相:甲醇-1%冰醋酸(60∶40),流速:1.0 mL/min,进样量:20μL,紫外检测器波长:254 nm,柱温:25℃。
1.2.2 混合对照品溶液 分别取对羟基苯甲酸甲酯和对羟基苯甲酸丙酯对照品,用流动相溶解并稀释成含量为0.04%和0.004%的单一对照品储备液。取0.04%的对羟基苯甲酸甲酯对照品储备液和0.004%的对羟基苯甲酸丙酯对照品储备液等体积混合,即得对羟基苯甲酸甲酯和对羟基苯甲酸丙酯含量分别为0.02%和0.002%的混合对照品储备液。取混合对照品储备液,制备系列浓度的混合对照品溶液,其中对羟基苯甲酸甲酯的系列浓度为0、0.002%、0.004%、0.006%、0.008%、0.010%和0.012%,对羟基苯甲酸丙酯的系列浓度为:0、0.000 2%、0.000 4%、0.000 6%、0.000 8%、0.001 0%和0.001 2%,按
1.2.1 项条件进样测定,以测定的峰面积为纵坐标,相应对照品浓度为横坐标,绘制标准曲线,得到回归方程。
1.2.3 供试品溶液 取重组人干扰素α2a注射液样品,用流动相进行10倍稀释,按1.2.1项条件进样测定,将峰面积代入标准曲线回归方程,计算样品中对羟基苯甲酸甲酯和对羟基苯甲酸丙酯含量。
1.2.4 供试品基质溶液 取不含对羟基苯甲酸甲酯和对羟基苯甲酸丙酯重组人干扰素α2a注射液空白制剂,用流动相进行10倍稀释,按1.2.1项条件进样测定。
1.3 方法的验证
1.3.1 系统适用性和专属性 分别取混合对照品溶液(对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的浓度分别为0.01%和0.001%)、供试品溶液和供试品基质溶液,按1.2.1项条件进样测定,分析待测成分色谱峰的理论塔板数、待测成分色谱峰与其他色谱法的分离度,比较不同进样的色谱图。
1.3.2 线性 取1.2.2项系列浓度的的混合对照品溶液,按1.2.1项条件进样测定,以测定的峰面积为纵坐标,相应对照品浓度为横坐标,进行线性回归,绘制标准曲线,计算R2值。
1.3.3 准确度 向供试品基质溶液中加入混合对照品溶液,充分混匀,使对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的含量分别为0.006%和0.000 6%,平行制备6份,按1.2.1项条件进样测定,计算对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的加标回收率。
1.3.4 重复性 取1批供试品溶液,连续进样6次,按1.2.1项条件进样测定,记录对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的峰面积,计算RSD。
1.3.5 耐用性 取1批供试品溶液,分别采用不同品牌的色谱柱:Phenomenex C18色谱柱(5μm,250 mm×4.6 mm)、Grace C18色谱柱(5μm,250 mm×4.6 mm)和Agilent C18色谱柱(5μm,250 mm×4.6 mm);不同的流动相甲醇-1%冰乙酸比例:55∶45、60∶40和65∶35;不同的柱温:25、30和35℃。按1.2项方法进行测定,考察试验条件变化对测定结果的影响。
1.4 气相色谱法 按《中国药典》三部(2020版)通则3116《对羟基苯甲酸甲酯、对羟基苯甲酸丙酯含量测定法》进行测定。
1.5 方法的初步应用 分别采用1.2和1.4项方法测定6批重组人干扰素α2a注射液中对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的含量。
1.6 统计学分析 应用Minitab软件,两种方法检测结果差异的比较采用配对t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 系统适用性和专属性 混合对照品溶液色谱图显示,对羟基苯甲酸甲酯和对羟基苯甲酸丙酯色谱峰的理论塔板数均大于9 000,待测成分色谱峰之间的分离度大于1.5,系统适用性良好;供试品基质溶液色谱图显示,未检出待测色谱峰;供试品溶液色谱图显示,2个待测成分(对羟基苯甲酸甲酯和对羟基苯甲酸丙酯)色谱峰的保留时间与混合对照品溶液的色谱图一致,待测成分色谱峰之间、待测成分色谱峰与其他色谱峰之间分离度均大于1.5,对待测成分的含量测定无干扰,方法的专属性良好。见图1。

图1 混合对照品溶液(A)、供试品溶液(B)和供试品基质溶液(C)色谱图Fig.1 Chromatograms of mixed reference(A),test sample(B)and base material(C)solutions
2.2 线性 对羟基苯甲酸甲酯浓度在0.002%~0.012%时,浓度与峰面积呈良好的线性关系,回归方程:y=1 204 693 661 x+45 682,R2为0.999 9;对羟基苯甲酸丙酯浓度在0.0002%~0.001 2%时,浓度与峰面积呈良好的线性关系,回归方程:y=1 043 508 571 x+9 771,R2为0.999 0。见图2。
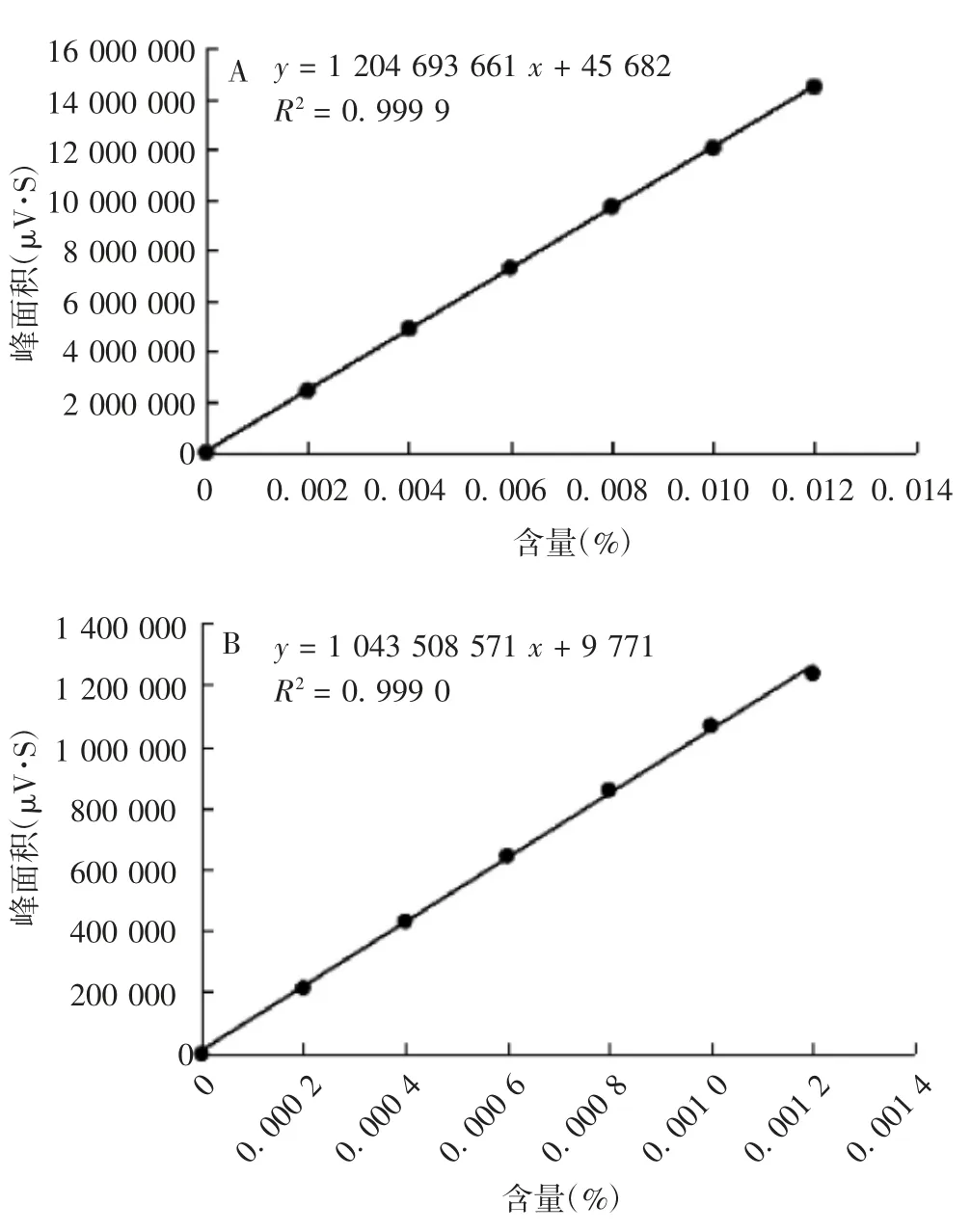
图2 对羟基苯甲酸甲酯(A)和对羟基苯甲酸丙酯(B)标准曲线Fig.2 Standard curve of methyl p-hydroxybenzoate(A)and propyl p-hydroxybenzoate(B)
2.3 准确度 对羟基苯甲酸甲酯6次加标回收率平均值为99.6%,RSD为0.13%;对羟基苯甲酸丙酯6次加标回收率平均值为99.2%,RSD为0.11%,方法准确度良好。见表1。
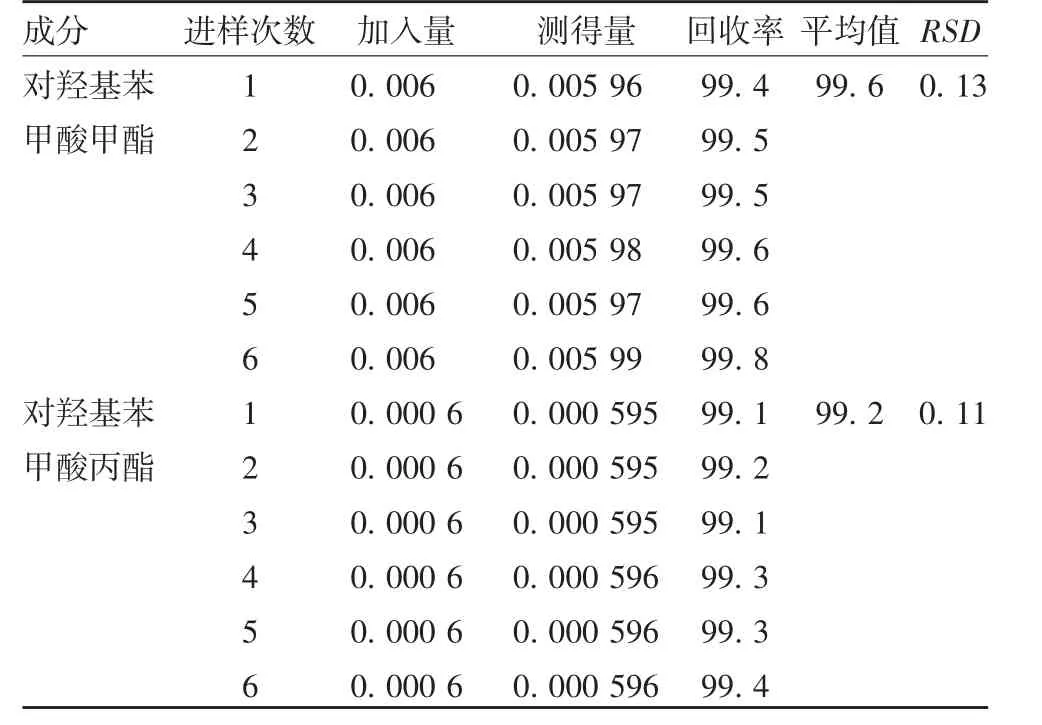
表1 加标回收率测定结果(%)Tab.1 Determination results of spike recovery(%)
2.4 重复性 连续进样6次,对羟基苯甲酸甲酯峰面积的RSD为0.05%,对羟基苯甲酸丙酯峰面积的RSD为0.07%,方法重复性良好,见表2。

表2 重复性测定结果Tab.2 Test for reproducibility
2.5 耐用性 在不同测定条件下,各色谱峰的峰型无显著变化,对羟基苯甲酸甲酯和对羟基苯甲酸丙酯含量测定结果的RSD均不超过2.0%,方法耐用性良好,见表3。
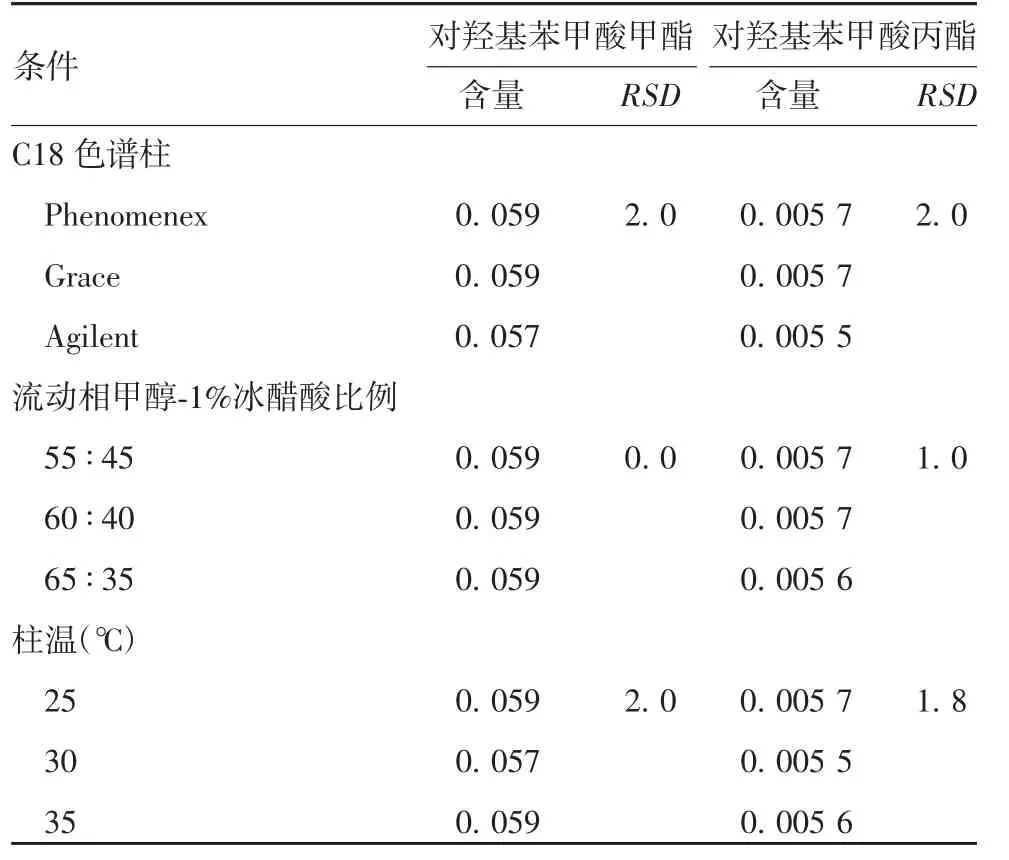
表3 耐用性测定结果(%)Tab.3 Test for durability(%)
2.6 方法的初步应用 对羟基苯甲酸甲酯含量的RP-HPLC法测定结果与气相色谱法相比差异有统计学意义(t=9.087,P<0.001),对羟基苯甲酸丙酯含量的RP-HPLC法测定结果与气相色谱法相比差异也有统计学意义(t=6.708,P<0.002)。对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的实际投料量分别为0.06%和0.006%,RP-HPLC法测定结果与投料量非常接近,而气相色谱法测定结果明显高于投料量。见表4。

表4 HPLC和气相色谱法测定结果对比(%)Tab.4 Determination results by HPLC and GC(%)
3 讨论
气相色谱法具有分离效率高、灵敏度高、选择性高等优点,主要用于分析气体和易挥发的有机物,进样方式包括直接进样和顶空进样[10]。气相色谱法用于对羟基苯甲酸甲酯和对羟基苯甲酸丙酯含量测定时,由于两者沸点较高,不易挥发,因此只能采用直接进样法。干扰素样品直接进样后,其中的蛋白和辅料容易污染色谱仪的隔垫、衬管和色谱柱[11],造成色谱图基线不稳定,色谱峰分叉,因此,进样2~3次后,需对色谱柱进行长时间的冲洗和平衡,隔垫、衬管等也需经常更换,这些均增加了检测时间和检测成本。另外,为了减少进样误差,《中国药典》三部(2020版)要求使用对苯二酚作为内标物。对苯二酚具有较强的毒性,对皮肤、黏膜有强烈的腐蚀作用,可抑制中枢神经系统或损害肝、皮肤功能,属于三类致癌物,影响操作人员健康,引起环境污染[12]。HPLC法具有灵敏度高、分离效率高、应用范围广等优点[13],HPLC系统受样品中蛋白和辅料的影响也较小,因此,本研究使用HPLC法代替气相色谱法测定人干扰素α2a注射液中对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的含量,由于对羟基苯甲酸甲酯和对羟基苯甲酸丙酯的疏水性不同,因此使用反相色谱柱进行分离。建立的HPLC法具有良好的专属性、线性、准确度、精密度和耐用性,与气相色谱法相比,具有操作简便、安全、准确度高的优势,可作为气相色谱法的替代方法。本方法的建立为《中国药典》方法的完善提供了技术支撑,也为其他蛋白药物中同类防腐剂含量的测定提供了参考。
猜你喜欢
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
云南化工(2020年11期)2021-01-14
百科知识(2016年22期)2016-12-24
当代化工研究(2016年5期)2016-03-20
化工进展(2015年3期)2015-11-11
中国当代医药(2015年10期)2015-03-01
海军医学杂志(2015年2期)2015-02-27
河北工业科技(2015年4期)2015-02-27
食品工业科技(2014年9期)2014-03-11
