
双环[4.2.1]⁃1(8)⁃壬烯的合成研究
2021-08-19 11:21朱珠杨鸿智
石油化工高等学校学报 2021年4期
朱珠,杨鸿智
双环[4.2.1]⁃1(8)⁃壬烯的合成研究
朱珠1,杨鸿智2
(1.辽宁石油化工大学 石油化工学院,辽宁 抚顺 113001;2.清华大学 药学院,北京 100084)
根据反Bredt规则,桥头双键化合物的烯烃应变能(Olefin Strain,OS)若小于71 kJ/mol,在室温条件下则可分离。双环[4.2.1]⁃1(8)⁃壬烯化合物的烯烃应变能小于71 kJ/mol,属于反Bredt规则分子。提出了一条新的合成路线,由2⁃环庚烯酮出发,以乙烯基溴化镁参与的1,4⁃共轭加成和分子内Wittig反应为关键步骤,以七步反应和17%总收率完成了双环[4.2.1]⁃1(8)⁃壬烯的克级规模合成。不同于以往的合成,本课题组采用的原料均商业可得,避免了原料自身的合成,简化了合成路线。此外,采用分子内Wittig反应作为关键反应,避免了异构体的生成。由于桥头双键化合物具有高张力和结构不稳定性,因而合成该类化合物具有很大的挑战性。但是,发展此类化合物的合成方法对于深入研究其化学性质和应用价值具有重要意义。
双环[4.2.1]⁃1(8)⁃壬烯; 乙烯基溴化镁; 1,4⁃共轭加成反应; 分子内Wittig反应
1902年,Bredt曾提出有机化学中的一个经验规律[1],含桥头双键结构的桥环化合物 (特殊大环除外)是不能稳定存在的,并以其名字命名,即通常所说的Bredt规则。然而,随着化学家对桥环化合物的深入研究,发现了一些反Bredt规则存在的分子,如:双环[4.2.1]⁃1(8)⁃壬烯化合物 (图1所示的化合物1)。根据Bredt规则,如果桥环化合物中的桥头碳原子上连有双键,则会受到双键烯烃角应变力的影响,使其结构不能稳定存在且化合物不可分离,因而关于合成桥环化合物的进一步研究并没有引起化学家的关注。直到20世纪80年代,科学家们尝试合成和分离含桥环双键的桥环化合物,重新解读了Bredt规则,引发了研究桥环双键化合物的热潮[2]。Maier和Schleyer两位化学家曾针对一些桥环双键化合物的烯烃应变能进行了分析总结,并提出了一些反Bredt规则[3]存在的分子(见图1)。(1)如果桥环双键化合物的烯烃应变能 (Olefin Strain,OS)小于71 kJ/mol,则在室温条件下,该类分子可以分离得到,并具有一定的稳定性,如图1所示的化合物1、2、3,校正了原Bredt规则中所述的该类化合物具有不可分离和不稳定的性质;(2)如果桥环双键化合物的烯烃应变能在71~88 kJ/mol,则可以在低温条件下观察到该类Bredt分子的存在,但在室温条件下不能分离得到,与在室温下该类桥环双键化合物不能稳定存在的结论一致;(3)如果某些Bredt分子的烯烃应变能大于88 kJ/mol,与第二类分子的稳定性相比,该类桥环双键化合物极其不稳定,只能在极低温度条件下观察到。由于含有桥头双键的化合物所具有的高张力和结构不稳定性,该类化合物的合成具有相当大的挑战性。从另一个角度考虑,发展此类化合物的合成方法对于深入研究其化学性质和应用价值具有重要意义。
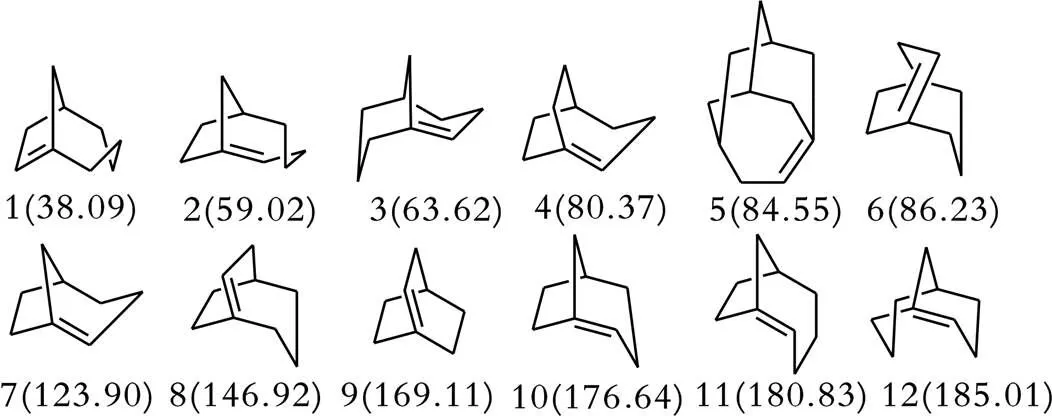
注:图中括号中数据为烯烃应变能(OS,kJ/mol)
本课题组综合考量了环张力和稳定性两个因素,最终选择双环[4.2.1]⁃1(8)⁃壬烯化合物作为合成目标。根据双环[4.2.1]⁃1(8)⁃壬烯分子的结构特点,设计了如图2所示的逆合成分析。

图2 双环[4.2.1]⁃1(8)⁃壬烯的逆合成分析
分析认为,目标产物双环[4.2.1]⁃1(8)⁃壬烯化合物1可以通过一步分子内的Wittig反应得到,而该反应的前体化合物2可以由化合物3脱除羰基保护基以及在三苯基膦的作用下得到。化合物3则可以通过前体化合物4经过溴代反应得到,后者可由化合物5通过羰基缩酮保护和双键的硼氢化⁃氧化反应得到,关键中间体5则可由商业廉价易得的环庚烯酮6通过1,4⁃共轭加成反应构建而成。
本文拟采用1,4⁃共轭加成和Wittig反应作为关键反应,由商业可得的环庚烯酮6经过七步反应实现了双环[4.2.1]⁃1(8)⁃壬烯化合物1的克级规模的制备。与已有合成方法相比较,本合成路线更为简洁高效,且中间体5也是双环[4.2.1]⁃1(8)⁃壬烯类衍生物[4]合成中常见的关键中间体,同时该策略也可用于其他类型的桥环双键化合物的合成研究,因此该路线具有广泛的参考应用价值。
1 实验部分
1.1 试剂与仪器
除非特殊说明,所有反应均在N2保护无水条件下进行,反应试剂均为购买后无需任何处理直接使用,所有化学试剂均为商业购得。
反应检测时使用的是青岛海洋化工所产的薄层色谱硅胶板(GF254),柱层析采用青岛海洋化工生产的200~300目硅胶。显色手段包括紫外、高锰酸钾 (加热)、磷钼酸(加热)和碘熏。
核磁谱图1H⁃NMR和13C⁃NMR采用Bruker AV400型核磁共振仪测定。使用氘代溶剂包括:氘代氯仿,其内残留的氯仿作为内标使用(核磁氢谱的化学位移为7.26,核磁碳谱的化学位移为77.16);多重峰采用以下简写:单峰(s),双重峰(d),三重峰(t),四重峰(q),宽峰(b),多重峰(m)。
1.2 合成步骤
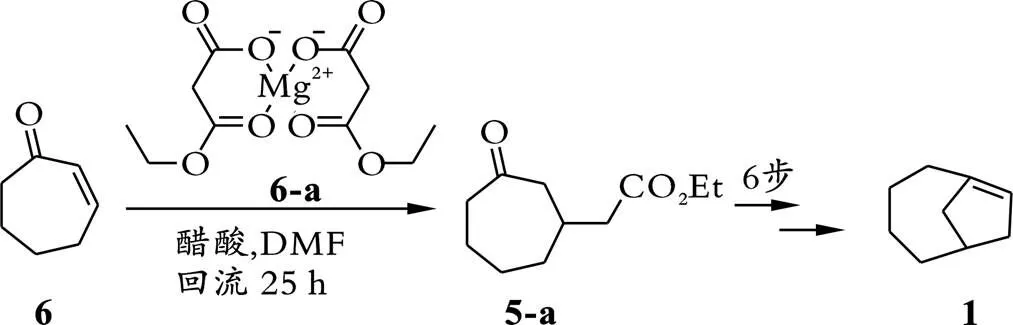
化合物5的合成[5](见式(1)):称取化合物CuI (1.7 g,8.9 mmol,0.1当量)于1 000 mL圆底烧瓶中,加入超干四氢呋喃溶液(425 mL),并加入大小适中的磁力搅拌子,将圆底烧瓶固定在磁力搅拌器上,用真空油泵抽空瓶中的空气,随后通入N2,重复三次前述换气步骤,保持瓶中无水无氧状态。将上述溶液冷却至-10 ℃,向该体系中缓慢加入乙烯基溴化镁(107.8 mL,107.8 mmol,1.0 mol/L的四氢呋喃溶液,1.2当量),滴加完成后保持该反应液在-10 ℃下继续搅拌1 h。随后将上述体系再冷却至-78 ℃,缓慢向该体系中滴加化合物6(10.0 mL,89.8 mmol,1.0当量)的四氢呋喃溶液(85 mL),继续保持在⁃78 ℃下搅拌1 h。TLC检测反应完全,加入饱和氯化铵水溶液淬灭反应,乙酸乙酯萃取(200 mL×3)上述混合液,合并有机相,无水Na2SO4干燥,抽滤,减压浓缩有机相,经制备板分离纯化得到化合物5(9.3 g,产率75%)。

化合物4⁃a[6]的合成(见式(2)):称取化合物5 (9.3 g,67.4 mmol,1.0当量)于250 mL圆底烧瓶中,加入甲苯溶液(83 mL),随后缓慢加入乙二醇(16.8 mL,296.5 mmol,4.4当量)和对甲基苯磺酸(83.0 mg,0.48 mmol,0.7%当量),并加入大小适中的磁力搅拌子,无水无氧操作同化合物5的合成。将上述反应液升温至120 ℃,加热回流15 h,用分水装置除水。TLC检测反应完全,加入2 g/L NaHCO3溶液淬灭反应,乙酸乙酯萃取(100 mL×3)上述混合液,合并有机相,无水Na2SO4干燥,抽滤,减压浓缩有机相,经制备板分离纯化得到化合物4⁃a(10.5 g,产率86%)。

化合物4[7]的合成(见式(3)):称取化合物4⁃a (10.5 g,57.7 mmol,1.0当量)于500 mL圆底烧瓶中,加入干燥的四氢呋喃溶液 (230 mL),并加入大小适中的磁力搅拌子,无水无氧操作同化合物5的合成。将上述溶液冷却至0 ℃,向该体系中缓慢滴加BH3.Me2S(43.3 mL,86.6 mmol,2.0 mol/L的四氢呋喃溶液,1.5当量),继续保持在0 ℃下搅拌0.5 h,随后移至室温下继续搅拌3.5 h。将上述反应混合物再次冷却至0 ℃,先后加入氢氧化钠溶液(28.9 mL,86.6 mmol,3 mol/L的水溶液,1.5当量)和过氧化氢溶液(25.9 mL,253.9 mmol,质量分数30%的水溶液,4.4当量)。将上述反应混合液缓慢升温至40 ℃,加热搅拌约1 h,TLC检测反应完全。加入饱和氯化铵溶液淬灭反应,乙醚萃取(100 mL×3)上述混合液,合并有机相,无水Na2SO4干燥,抽滤,减压浓缩有机相,经制备板分离纯化得到化合物4(9.5 g,产率82%)。

化合物3[8]的合成(见式(4)):称取化合物4 (9.5 g,47.5 mmol,1.0当量)于500 mL圆底烧瓶中,加入超干二氯甲烷溶液(238 mL),并加入大小适中的磁力搅拌子,无水无氧操作同化合物5的合成。先后向上述体系中加入CBr4(19.7 g,59.4 mmol,1.3当量)和PPh3(18.7 g,71.3 mmol,1.5当量),将上述反应液缓慢升温至50 ℃加热回流20 h。TLC检测反应完全,将上述反应液冷却至室温,加入质量分数5%NaHCO3溶液淬灭反应,乙酸乙酯萃取(40 mL×3)上述混合液,水洗(50 mL×3),合并有机相,无水Na2SO4干燥,抽滤,减压浓缩有机相,经制备板分离纯化得到化合物3(8.6 g,产率69%)。

化合物2⁃a[9]的合成(见式(5)):称取化合物3 (8.6 g,32.8 mmol,1.0当量)于500 mL圆底烧瓶中,加入超干四氢呋喃溶液(60 mL),并加入大小适中的磁力搅拌子,无水无氧操作同化合物5的合成。随后缓慢滴加质量分数10%硫酸溶液(238 mL)。将上述反应液升温至80 ℃,加热回流5 h。TLC检测反应完全,向上述反应混合物中加入氢氧化钠溶液,将pH调至7~8。乙酸乙酯萃取(100 mL×3)上述混合液,合并有机相,无水Na2SO4干燥,抽滤,减压浓缩有机相,经制备板分离纯化得到化合物2⁃a(6.6 g,产率92%)。

化合物2的合成[10](见式(6)):称取化合物2⁃a (6.6 g,30.3 mmol,1.0当量)于25 mL圆底烧瓶中,加入超干甲苯溶液(7 mL),向反应体系中缓慢加入白色固体PPh3[11](8.7 g,33.3 mmol,1.1当量),并加入大小适中的磁力搅拌子,无水无氧操作同化合物5的合成。将上述反应体系缓慢升温回流24 h,TLC监测反应完全,反应完全后冷却至室温,将反应体系的上层溶液倾倒出去,并重新加入超干的甲苯溶液(8 mL),再次回流20 min,将上述步骤重复两遍。之后,将下层反应相进行真空旋蒸浓缩,并真空干燥12 h,得到白色固体化合物2的粗品。化合物2不进行纯化直接投入下一步反应。
双环[4.2.1]⁃1(8)⁃壬烯化合物1的合成[6](见式(7)):在一个体积为250 mL的三口圆底烧瓶中加入大小适中的磁力搅拌子,无水无氧操作同化合物5的合成。称取已用戊烷洗涤过的氢化钠(1.7 g,72.7 mmol,2.4当量)于250 mL三口圆底烧瓶中。将上一步合成出的粗品化合物2用四乙二醇二甲醚溶液(102 mL)溶解,并缓慢加入到圆底烧瓶中,继续缓慢滴加2⁃甲基⁃2⁃丁醇(3.3 mL,30.0 mmol,1.0当量),滴加完成后将反应液升温至70 ℃,并在该温度下加热搅拌30 min;之后,反应体系在4 h内缓慢升温至120 ℃。TLC监测反应进程,反应完全后,将反应体系置于冰浴中,并减压浓缩得到化合物1的粗品,将该粗品经制备板分离纯化,得到纯的化合物1(1.9 g,产率52%)。


2 结果与讨论
2.1 与已有合成路线的比较
综上所述,以商业可得的环庚烯酮6为起始原料,经过七步简单的转化,最终以17%的总收率完成了双环[4.2.1]⁃1(8)⁃壬烯化合物1的合成。通过核磁1H⁃NMR和13C⁃NMR对化合物1进行结构表征,与已报道的核磁数据完全一致[12]。早在1977年,K. B.Becker课题组[6]开展了双环[4.2.1]⁃1(8)⁃壬烯1的合成研究,其关键反应虽然也是分子内的Wittig反应,但其中镁络合物6⁃a需要通过两步反应合成[13],见式(8)。即:首先以丙二酸单乙酯钾盐为原料,在盐酸的加入下生成丙二酸单乙酯化合物;而后,将镁粉加入到含丙二酸单乙酯化合物的乙醇溶液中;经过以上两步反应最终得到镁络合物6⁃a。与K.B.Becker课题组的合成工作相比,本课题组直接从商业可得的乙烯基溴化镁出发,与环庚烯酮发生1,4⁃共轭加成反应,简化了合成路线,避免了原料自身的合成过程。除此之外,各种官能团化的双环[4.2.1]⁃1(8)⁃壬烯衍生物,都可以通过关键中间体5的衍生化得到;而中间体5的衍生化,只需带有不同取代基的商业可得的乙烯基溴化镁和环庚烯酮发生1,4⁃共轭加成反应构建而成。此外,不同于分子内 Wittig的关键反应,R.Keese[14]曾采用霍夫曼消除[15]合成双环[4.2.1]⁃1(8)⁃壬烯化合物,遗憾的是,并没有得到单一的双环[4.2.1]⁃1(8)⁃壬烯,而是桥环双键处于七元环和五元环上的异构体混合物,由于该异构体理化性质非常相似,造成了分离纯化的困难,因而该策略并不是理想的合成手段。与前人合成双环[4.2.1]⁃1(8)⁃壬烯工作相比,虽然本研究步骤为7步,但所采用的原料均商业可得,简化合成过程,提高合成效率,该合成路线具有更高应用价值。
(8)
2.2 化合物1和5的结构表征
化合物5的核磁1H⁃NMR和13C⁃NMR如图3所示。由图3可见,1H⁃NMR (400 MHz,CDCl3),5.70为m峰,1H;4.89为m峰,2H,这是中间体5的环外侧链双键的特征峰,是判断化合物5成功合成的最主要特征峰。2.41处为m峰,4H,这是羰基α位的特征峰。2.29处为m峰,1H,这是双键α位的特征峰。1.84处为m峰,3H;1.53处为m峰,1H;1.35处为m峰,2H。13C⁃NMR (100 MHz,CDCl3),碳原子的依此是213.32、142.48、112.80、48.90、43.92、39.70、36.58、28.29、23.98。综合以上各项数据,证明已经成功合成关键中间体化合物5。

图3 化合物5的1H⁃NMR(CDCl3,400 MHz)和13C⁃NMR (CDCl3,100 MHz)
化合物1的核磁1H⁃NMR和13C⁃NMR如图4所示。由图4可见,1H⁃NMR (400 MHz,CDCl3),5.47为s峰,1H,这是双环[4.2.1]⁃1(8)⁃壬烯的桥头双键上的氢,是判断化合物1成功合成的最主要特征峰。2.68处为m峰,1H;2.40处为m峰,2H;2.22处为m峰,1H;2.06处为m峰,1个H;1.70处为m峰,4H;1.45为m峰,2H;1.16处为m峰,1H;1.04处为m峰,1H;13C⁃NMR (100 MHz,CDCl3),碳原子的依此是147.28、127.30、44.98、38.29、34.40、32.00、26.61、23.49。综合以上各项数据,证明已经成功合成双环[4.2.1]⁃1(8)⁃壬烯化合物1。
在后续的研究工作中,将双环[4.2.1]⁃1(8)⁃壬烯化合物作为合成砌砖,利用其自身具有的高反应活性的特性,进一步展开一系列的基于其结构中活性烯烃的亲电加成反应的研究[16]。
3 结 论
从环庚烯酮出发,以乙烯基溴化镁参与的1,4⁃共轭加成和分子内Wittig反应为关键步骤,以七步反应和17%总收率完成了双环[4.2.1]⁃1(8)⁃壬烯的克级规模合成。

图4 化合物1的1H⁃NMR(CDCl3,400 MHz)和13C⁃NMR (CDCl3,100 MHz)
[1]Lee G A,Lin H C.Synthesis of an anti⁃Bredt compound,bicyclo[3.2.2]nona⁃1,6,8⁃triene,via the isomerization of tricyclo[3.2.2.02,4]nona⁃2,6⁃diene[J].Org. Lett.,2014,16(20):5275⁃5277.
[2]Becker K B,Chappuis J L.Synthesis of 1⁃bicyclo[3.2.2]nonene and 1(7)⁃bicyclo[3.2.2]nonene by intramolecular Wittig reaction[J].Helvetica Chimica Acta,1980,63(7):1812⁃1822.
[3]Bly R S,Bly R K,Hossain M M,et al.[Fp⁃.eta.1⁃(1⁃polycycloalkyl) methylidene]+to [Fp⁃.eta.2⁃(1⁃homopoly⁃cycloalkene)+rearrangement.Carbon migration in iron (II) alkylidenes.A new route to stabilized bridgehead olefins[J].J.Am. Chem. Soc.,1988,110(23):7723⁃7730.
[4]Carruthers W,Qureshi M I.Bredt’s rule.A derivative of bicyclo[4.2.1]non⁃1(8)⁃ene[J].J.Chem.Soc.D,1969(14):832⁃833.
[5]Cheng H M,Tian W,Peixoto P A,et al.Synthesis of ent⁃nanolobatolide[J].Angew. Chem. Int. Ed.,2011,50(18):4165⁃4168.
[6]Becker K B.The synthesis of strained methylene⁃bridged bicyclic olefins by the intramolecular Wittig reaction[J].Helvetica Chimica Acta,1977,60(1):81⁃93.
[7]Piers E,Gavai A V.A(Z)⁃ethylidenecyclopentane annulation method.Total syntheses of (±)⁃anhydrooplopanone,(±)⁃oplopanone,and (±)⁃8⁃epi⁃oplopanone[J].J. Org. Chem.,1990,55(8):2380⁃2390.
[8]Ravina E,Negreira J,Masaguer C F,et al.Conformationally constrained butyrophenones with mixed dipaminergic (D2) and serotoninergic (5⁃HT2A,5⁃HT2C) affinities:Synthesis,pharmacology,3D⁃QSAR,and molecular modeling of (aminoalkyl) benzo⁃and thienocycloalkanones as putative atypical antisychotics[J].J. Med. Chem.,1999,42(15):2774⁃2797.
[9]Mishura A,Sklyarova A,Sharapa D,et al.Stereoselective preparation of mono⁃and bis⁃derivatives of pentacyclo[6.3.0.02,6.03,10.05,9]undecane (D3⁃trishomocubane)[J].Cent. Eur. J. Chem.,2013,11(12):2144⁃2150.
[10] Nicolaou K C,Yue E W,Greca S L,et al.Synthesis of zaragozic acid A/Squalestatin S1[J].Chem. Eur. J. ,1995,1(7):467⁃494.
[11] 陈阳,孙京,周明东.(1⁃丁基⁃3,3⁃二苯基丙二烯基)二苯基氧膦的合成[J].辽宁石油化工大学学报,2017,37(3):6⁃10.Chen Y,Sun J,Zhou M D.Synthesis of 1,1⁃diphenylhepta⁃1,2⁃dien⁃3⁃yl diphenyl phosphine oxide compound[J].Journal of Liaoning Shihua University,2017,37(3):6⁃10.
[12]Wiseman J R,Chan H F,Ahola C J.Bredt’s rule.II.Synthesis of bicyclo[4.2.1]non⁃1(2)⁃ene and bicyclo[4.2.1]non⁃1(8)⁃ene[J].J.Am.Chem.Soc.,1969,91(10):2812⁃2813.
[13] Mcmurry J E,Andrus W A,Musser J H.Conjugate addition⁃decarboxylation of magnesium monoethyl malonate to enones:Aconvenient one⁃flask addition of acetata[J].Synthetic Communications,1978,8(1):53⁃57.
[14] Keese R.Methods for the preparation of bridgehead olefins[J].Angew. Chem. Int. Ed.,1975,14(8):528⁃538.
[15]Becker K B,Pfluger R W.A facile synthesis of strained bridgehead olefins by a gas⁃phase elimination reaction[J].Tetrahedron Letters,1979,20(39):3715⁃3716.
[16] Becker K B.Electrophilic additions to strained bridgehead olefins.Estimation of strain by comparison with the solvolysis of bridgehead bromides[J].Helvetica Chimica Acta,1977,60(1):94⁃102.
Synthesis of Bicyclo[4.2.1]⁃1(8)⁃Nonylene
Zhu Zhu1,Yang Hongzhi2
(1.School of Petrochemical Technology,Liaoning Petrochemical University,Fushun Liaoning 113001,China;2.School of Pharmaceutical Sciences,Tsinghua University,Beijing 100084,China)
According to anti⁃Bredt rule,if olefin strain (OS) of alkene with double bond in bridgehead is less than 71 kJ/mol,it can be isolated at room temperature.The OS of bicyclo[4.2.1]⁃1(8)⁃nonylene is less than 71 kJ/mol,conforming with anti⁃Bredt rule.A novel synthetic route of gram⁃scalable bicyclo[4.2.1]⁃1(8)⁃nonylene was reported,which was synthesized in seven steps with 17% overall yield,characterized by starting from 2⁃cycloheptenone using a vinyl magnesium bromide⁃mediated 1,4⁃conjugation as addition and an intramolecular Wittig reaction as the key steps.Different from the previous synthesis,the raw materials used in this method are all commercially available,which avoids the synthesis of raw materials themselves and simplifies the synthesis route.In addition,intramolecular Wittig reaction is used as the key reaction to avoid the generation of isomers.It is very challenging to synthesize alkene with double bond in bridgehead because of their high strain energy and instability.However, development of synthetic routes for such compounds is of great significance for further study of their chemical properties and application.
Bicyclo[4.2.1]⁃1(8)⁃nonylene; Vinyl magnesium bromide; 1,4⁃Conjugation addition; Intramolecular Wittig reaction
TE624
A
10.3969/j.issn.1006⁃396X.2021.04.002
1006⁃396X(2021)04⁃0009⁃05
http://journal.lnpu.edu.cn
2020⁃04⁃12
2021⁃06⁃10
朱珠(1995⁃),女,硕士研究生,从事天然产物全合成以及药物化学等研究;E⁃mail:737007550@qq.com。
杨鸿智(1989⁃),女,博士研究生,从事天然产物全合成以及药物化学等研究;E⁃mail:yhz17@mails.tsinghua.edu.cn。
(编辑 闫玉玲)
猜你喜欢
科学导报(2022年41期)2022-07-13
安徽农学通报(2022年8期)2022-05-06
宝钢技术(2022年1期)2022-03-14
油气·石油与天然气科学(2021年12期)2021-12-11
——人-时间资料率比分析与SAS实现
四川精神卫生(2021年4期)2021-09-10
合成树脂及塑料(2020年4期)2020-09-20
石油化工(2020年2期)2020-04-04
黑龙江交通科技(2020年5期)2020-01-13
分析化学(2017年12期)2017-12-25
家用汽车(2016年12期)2017-02-09
