
钾掺杂对Fe/GO催化合成气制α-烯烃的影响
2021-08-15 07:51李玉峰杨鹏举胥月兵刘小浩
燃料化学学报 2021年7期
李玉峰,杨鹏举,姜 枫,刘 冰,胥月兵,刘小浩
(化学与材料工程学院 江南大学,江苏 无锡 214122)
费托合成(FTS)技术能够将以煤、天然气、生物质来源的合成气转化为高附加值的化学品和清洁燃料[1,2],为解决人类社会可持续发展与环境污染问题提供了重要保障。铁基和钴基催化剂是常用的FTS 催化剂,一直以来受到研究人员的广泛关注[3−15]。由于铁基催化剂更弱的加氢反应能力,相比于钴基催化剂更有利于α-烯烃的生成[16−22]。此外,铁基催化剂表现出更好的抗硫和氯等强给电子体中毒的能力,因此,铁基催化剂是合成气制α-烯烃最有应用前景的催化剂[23,24]。然而,铁基费托催化剂具有容易积炭而失活、反应过程中铁物相演变复杂[25,26]、高CO2选择性[27,28]等缺点。因此,如何利用铁基催化剂获得高α-烯烃选择性,同时具有良好的催化稳定性和低的二氧化碳选择性是极具挑战性的研究课题。
与传统的氧化物载体(SiO2,γ-Al2O3等)相比较,铁与碳材料之间具有弱相互作用并且不会发生固相反应生成不可还原的铁物相,相反,其还能促进铁氧化物的还原,因此,碳材料广泛应用于铁基催化剂的制备和费托合成反应。然而,由于铁与碳载体之间弱的相互作用,反应过程中铁纳米粒子容易长大导致催化剂活性下降。Galvis 等[17]利用碳纳米纤维(CNF)做载体负载Fe 制备催化剂,并用S 和Na 助剂对催化剂进行改性,获得高达60%的低碳烯烃选择性。Chen 等[29]制备了碳纳米管限域的铁基催化剂(Fe-in-CNT),CNT 的限域作用能显著抑制反应过程中Fe 纳米粒子生长,且该催化剂相比非限域催化剂(Fe-out-CNT)具有更高的催化活性。此外,多孔炭、MOF 衍生炭等炭包覆型铁基催化剂均有文献报道[30]。氧化石墨烯(GO)是一种二维碳材料,含有丰富的含氧官能团和独特的电子性质[31],这些优点有利于制备高性能的负载型铁基FTS 催化剂。Wei 等[32]将不同Fe 前驱体负载到GO 中,得到Fe 粒径在5−80 nm的Fe/GO 催化剂。其中,粒径集中在10−50 nm 的宽范围具有较好的催化性能。不同于上述文献报道,本课题组开发了一种新型的铁基费托催化剂制备方法:将粒径约为120 nm 的α-Fe2O3纳米粒子与GO 简单混合,并在FTS 条件下对铁催化剂的演变过程进行了深入研究[33]。相比其他碳材料(活性炭、PVP 和PAA),α-Fe2O3仅在GO 存在下表现出独特的演变行为并形成了高性能的费托合成制α-烯烃催化剂。具体而言,大颗粒α-Fe2O3逐渐演变成小尺寸的圆柱状纳米胶囊碳化铁,其平均直径约为9.6 nm。从HRTEM 照片中可以看出,纳米胶囊碳化铁粒子呈现单一的χ-Fe5C2(510)晶面,其外层为约2 nm 厚的无定型炭层,这种包覆结构能有效抑制纳米粒子烧结,具有优异的活性、催化稳定性和高α-烯烃选择性。与此同时,CO2的选择性也高达45%−48%。助剂对铁基费托合成具有重要调节作用。K[19]、Na[34]、Mn[35]、S[17]、N[18]等助剂添加可提高铁基费托合成中烯烃选择性。助剂的添加也会影响碳化铁物相、调节活性中心电子性质,进而改变产物分布。
本实验进一步研究了GO 和K 助剂共存时大颗粒铁纳米粒子的演变行为。不同于以前的研究,本实验利用大尺寸Fe3O4微球替代α-Fe2O3纳米粒子。Fe3O4微球平均粒径约为580 nm,由平均粒径约为30 nm Fe3O4纳米晶粒聚集而成。通过本研究期望达到以下目标:揭示GO 和K 对Fe3O4演变及其物相的影响;保持高α-烯烃选择性的前提下,明显降低CO2选择性。
本研究采用水热法合成了规整的大尺寸Fe3O4微球。在FTS 反应条件下,考察了GO、K 助剂以及H2/CO 比对于Fe3O4演变、催化活性和产物选择性的影响。采用BET、XRD、Raman 光谱、H2-TPR、CO-TPD、SEM、TEM 和穆斯堡尔谱对反应前后催化剂进行了表征。
1 实验部分
1.1 催化剂的制备
采用水热法制备Fe3O4微球[36]。方法如下:将7.27 g Fe(NO3)3·9H2O 溶解在180 mL 乙二醇中形成溶液,然后加入12.96 g NaAc·3H2O。搅拌30 min后,将混合溶液加入含有聚四氟乙烯内衬的水热釜中,并加热至200℃,保持12 h。冷却至室温后,产物分别用去离子水和乙醇各洗涤几次,然后在60℃下干燥12 h。利用浸渍法制备K 掺杂的Fe3O4微球。将KNO3溶液浸渍到上述合成的Fe3O4微球中。浸渍后的样品在120℃下干燥12 h,随后在300℃下焙烧2 h。K 掺杂的催化剂表示为xKFe,其中,x 代表金属K 与Fe 的质量比。
Fe3O4/GO 催化剂采用如下方法制备:首先采用改进Hummer 方法制备GO[37],随后分别将Fe3O4微球与GO 在水中超声分散得到悬浮液,并将两悬浮液混合、超声、离心和干燥,即得到催化剂。其中,Fe3O4与GO 的质量比为1∶1,催化剂命名为Fe/GO。对于xKFe 催化剂,将其与GO 按同样方法混合,所得催化剂命名为xKFe/GO,其中,Fe3O4与GO 的质量比为1∶1,x 代表K 与Fe 的质量比。
1.2 FTS 反应
FTS 反应在内径为12 mm 的高压固定床反应器中进行。在反应之前,将100 mg 催化剂用2 g石英砂稀释,装入到反应器中。采用37 mL/min的H2在350℃和0.2 MPa 下还原3 h,升温速率为2℃/min。随后在H2气氛下床层冷却至280℃,接着将H2/CO/N2物质的量比为48.5∶48.5∶3 的合成气切入反应器,系统压力升至1 MPa,床层温度逐渐升高至340℃开始反应。合成气的流量设定为37 mL/min,反应空速为22.2 L/(g∙h),反应时间为50 h。
采用集成双检测器气相色谱仪(Agilent GC 7820A)对气相产物进行在线分析。其中,热导检测器(TCD)用于分析CO2、H2、CO、N2和CH4;火焰离子化检测器(FID)用于分析C1−C7碳氢化合物的组成。反应的液体产品和蜡分别在0℃和120℃下通过冷阱、热阱收集,采用气相色谱仪(岛津GC-2014,HP-1 毛细管柱)进行离线分析。根据摩尔数计算CO 转化率和产物选择性。碳平衡为95%−103%。
1.3 催化剂的表征
N2吸附-解吸等温线采用美国康塔公司AutosorbiQ 仪器测定。在分析之前,150 mg 催化剂在200℃下真空脱气4 h。比表面积采用Brunauer-Emmett-Teller(BET)法计算。孔径和孔体积采用Barrett-Joyner-Halenda(BJH)法并根据N2解吸等温线计算。新鲜催化剂的形貌和尺寸采用日本日立公司S-4800 场发射扫描电子显微镜(SEM)测定,加速电压为2.0 kV。新鲜催化剂和反应后催化剂的微观结构采用日本电子JEM-2100 型透射电子显微镜(TEM)测定,加速电压为200 kV。催化剂的X 射线衍射(XRD)谱图采用布鲁克AXS-D2 型衍射仪进行分析测定,辐射源为Cu Kα(λ=0.15418 nm),扫描步长为0.02°,为5°−90°扫描。Raman 光谱采用英国雷尼绍公司inVia 显微共聚焦拉曼光谱仪测定,激光器波长为532 nm,功率为50 mW。催化剂 的57Fe 穆斯堡尔(Mössbauer)谱采用WISSEL 1550 穆斯堡尔机电光谱仪测定,采用Pd 为基体的57Co 为辐照源,25 μm α-Fe 箔进行速度标定,异构体位移(IS)参照α-Fe。采用MossWinn 3.0i 程序根据最小二乘法对光谱拟合为单峰、四重峰和磁性六重态,由吸收峰面积推导出催化剂的物相组成。H2-TPR 采用日本MircrotraceBEL 公司全自动化学吸附分析仪测定,检测器为TCD,并采用质谱对出口气体进行定性分析。首先,将50 mg 样品装入石英管中,在200℃下用N2吹扫1 h 后冷却至室温;随后通入10%H2/Ar(30 mL/min),以10℃/min 升温速率升至900℃,记录H2消耗曲线。COTPD 在相同仪器上进行测定。首先,将150 mg 样品装入石英管中,在350℃下用10%H2/N2(30 mL/min)还原3 h 后,切入N2冷却至室温;然后通入5%CO/N2(30 mL/min)持续1 h;随后采用He(30 mL/min)吹扫催化剂30 min 除去弱吸附物种,并在He 气氛中以10℃/min 的升温速率升至900℃,记录脱附曲线。
2 结果与讨论
2.1 新鲜催化剂的表征
新鲜催化剂的XRD 谱图和N2吸附-脱附曲线分别如图1、图2 所示。Fe3O4微球在30.4°、35.8°、43.5°、57.5°和63.1°处出现典型的Fe3O4(JCPDS-75-499)衍射特征峰,表明其结晶良好,计算得平均晶粒粒径为30.8 nm。催化剂的SEM 照片如图3所示,可观察到Fe3O4微球颗粒形貌规整,其平均直径约为580 nm,表明Fe3O4微球由小颗粒纳米晶粒堆积而成[38]。晶粒堆积导致Fe3O4微球具有低比表面积(6.7 m2/g)和低的孔容(图2)。K 的添加导致比表面积略有减少,在XRD 谱图中未观察到K 物种存在。对于Fe/GO 催化剂,在2θ=12.2°处的衍射峰归属为GO 的C(002)晶面[39]。此外,SEM照片中可以看出,GO 和K 修饰的Fe3O4微球仍保持其原来形貌。
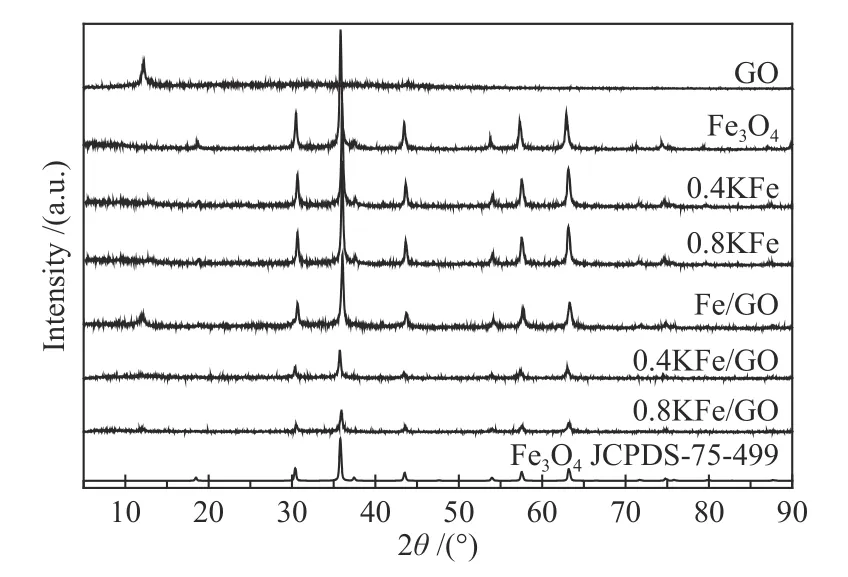
图1 催化剂的XRD 谱图Figure 1 XRD patterns of the various fresh catalysts
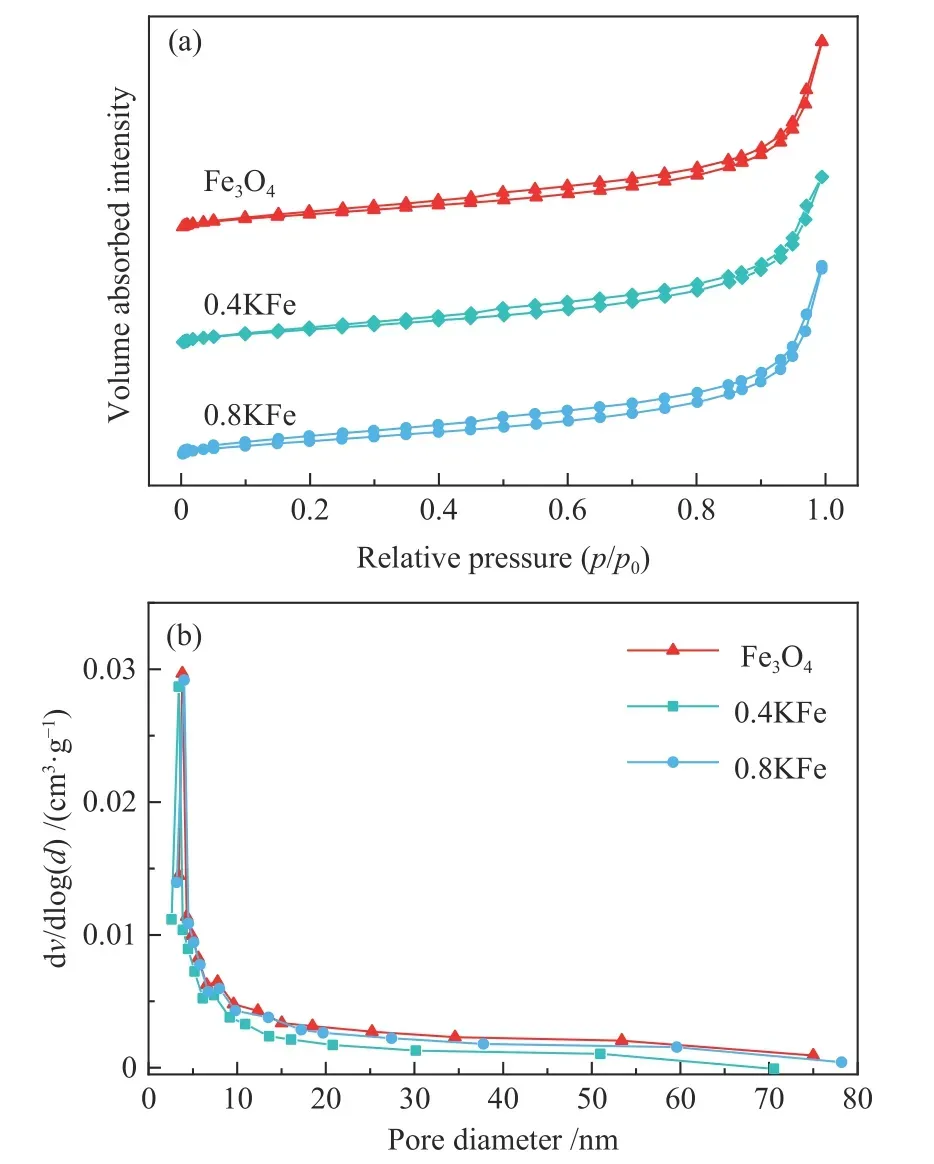
图2 新鲜催化剂的N2 吸附-脱附曲线(a)及相应的孔径分布(b)Figure 2 N2 adsorption-desorption isotherms (a) and corresponding pore size distribution curves (b)

图3 新鲜催化剂的SEM 照片Figure 3 SEM images of the fresh catalysts
为了进一步研究GO 的性质,对催化剂进行了拉曼光谱表征。如图4(a)所示,GO 的拉曼光谱显示出典型的G 带(1586 cm−1)和D 带(1325 cm−1)峰[40],D 与G 的强度比约为1.17。G 带源自于sp2C 原子的E2g振动,而D 带是由于局部缺陷,特别是位于边缘的C 原子的缺陷结构的A1g振动产生[41]。通常D/G 是石墨烯结构量度的重要参数,对于高度有序的GO,该比值接近于零。相比于纯GO,Fe/GO 催化剂的D/G 强度明显增加,表明Fe3O4微球的引入导致缺陷碳位点数量增加。载体中的缺陷位点有利于锚定铁纳米粒子,阻止其迁移和聚集[42]。在Fe3O4微球中也观察到了C 原子D 带和G 带振动峰,这可能是由于Fe3O4制备过程中有机物分解时残留了少量炭杂质。此外,Fe3O4在660 cm−1处的特征拉曼散射峰归因于其Alg振动和O 原子沿Fe−O 键的对称拉伸[43]。该结果表明,Fe 物种以Fe3O4形式存在,与XRD 结果一致。如图4(b)的CO-TPD 谱图所示,单独的Fe3O4或GO 显示弱的CO 解吸峰,而两者的结合不仅提高了CO 解吸温度,而且还明显提高了CO 的吸附量。这表明GO 的引入有利于CO 的吸附和活化。
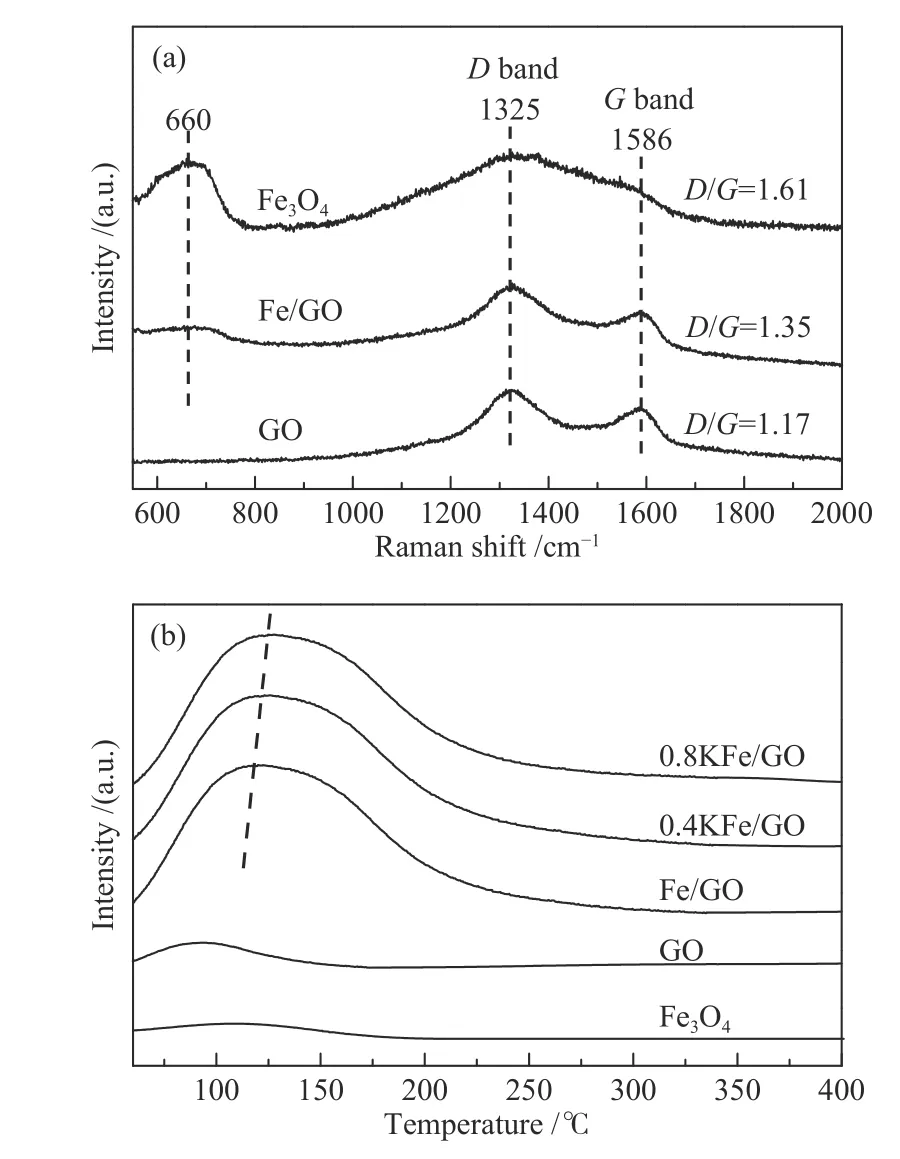
图4 新鲜催化剂拉曼光谱(a)和CO-TPD 谱图(b)Figure 4 Raman spectra (a) and CO-TPD (b) of the fresh catalysts
随着K 含量的增加,CO 解吸温度也逐渐提高,表明K 有利于CO 的吸附强度增加,有利于其活化。Cheng 等[19]发现,提高KFe/rGO 催化剂中K 的含量,CO 解吸温度也会相应提高,这与本实验结果一致。在Fe 基催化剂中,碱金属可向Fe 转移电子,提高Fe 电子云密度,从而增强Fe 中d 电子对CO 反键轨道的反馈作用,促进CO 的解离[19,21,22]。由此可以看出,GO 和K 的添加均有利于CO 的解离,提高催化剂表面C*物种浓度,根据质量作用定律,C*物种浓度的提高将有利于偶联反应的进行(见下文)[44]。
催化剂的H2-TPR 谱图如图5(a)所示,相应的定量数据分析如表1 所示。由图5(a)可以看出,Fe3O4的还原分为两个阶段。第一阶段在400−500℃,归因于表面氧化形成的Fe2O3还原为Fe3O4;第二阶段在500−730℃,对应于Fe3O4经FeO 还原形成金属Fe。在GO 修饰后,由于GO 中的含氧官能团与Fe 物种的相互作用,第一阶段还原受到抑制[42],第二阶段还原温度稍有降低,但还原峰也相应减少,表明GO 加入抑制了Fe3O4的还原。750℃以上的信号峰归因于GO 载体的分解气化,因为含氧基团(例如羧基)可以在H2中分解释放出CO[18,45]。K 助剂添加导致Fe2O3还原峰增大,且峰的位置出现在更高的温度。这可能是由于制备K/Fe 催化剂时需在300℃空气中焙烧2 h,导致少量Fe3O4氧化为Fe2O3。还原峰的后移可能归因于K 对催化剂还原的抑制作用。在500−560℃处观察到一个新峰,这是由于KNO3分解生成NO2。在以前的研究中,以NaNO3为前驱体制备的Na/SiO2催化剂也表现出相似的H2-TPR 谱图[46]。为了确认这一结论,以KNO3为前驱体制备了K/SiO2催化剂,其中,K 含量为1%,在同样测定条件下进行H2-TPR 实验,含K 样品均在相同位置出现了H2-TPR峰,说明该峰源于KNO3分解。图5(b)中的质谱信号证实了该峰对应KNO3分解形成NO2。从图5(a)中可以看出,K 助剂的加入使Fe3O4的主要还原峰移至更高的温度,这可能归因于碱金属K 抑制了催化剂表面H2分子的解离。文献研究表明,碱金属助剂有利于稳定H2异裂解离形成的H−,抑制了电子从H−向金属阳离子转移,进而使H2O 的形成具有更高的能垒[42]。根据730℃之前的TPR 峰面积计算每克催化剂消耗H2的摩尔量,并将其与理论值相比较得到催化剂的还原度(见表1)。其中,含K 催化剂的计算结果是除去了NO2对TCD 信号峰面积的贡献。结果表明,K 的加入提高了催化剂的还原度。在以前的研究中,本课题组采用氧空位形成能来评估氧化铁的还原行为[22],计算得到Na2O/Fe3O4的氧空位形成能为1.73 eV,明显低于纯Fe3O4(6.16 eV),表明Na 的加入在热力学上有利于氧化铁的还原。这与本研究的实验结果是一致的。此外,0.4KFe/GO 和0.8KFe/GO 催化剂的H2-TPR 结果表明,K 的添加能促进GO 在高温下的分解。

图5 催化剂的H2-TPR 谱图Figure 5 H2-TPR profiles of catalysts
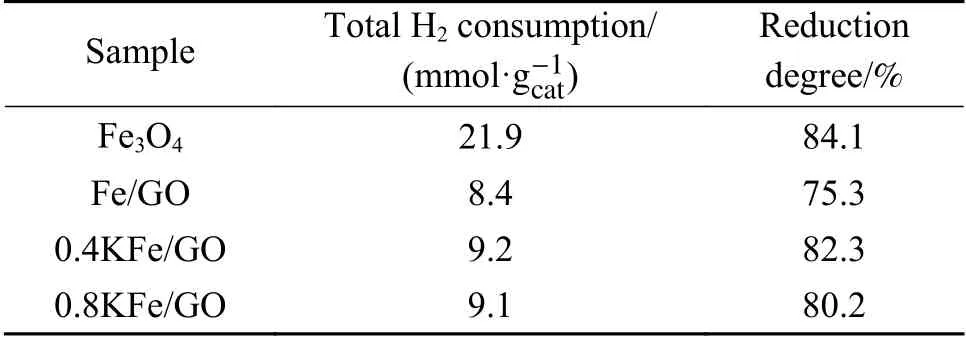
表1 新鲜催化剂的H2-TPR 表征Table 1 H2-TPR results of the fresh catalysts
2.2 反应后催化剂的表征
在FTS 反应条件下Fe3O4和Fe/GO 的形貌演变如图6 所示。由图6 可知,两种催化剂表现出完全不同的演变行为。对于纯Fe3O4,反应后催化剂表现出严重的积炭和粒子烧结,积炭层的厚度高达21 nm,铁纳米粒子的尺寸也从反应前的30.8 nm增长到74 nm(图6(b))。如图6(d)所示,Fe/GO 呈现出完全不同的演变过程,大尺寸的Fe3O4颗粒演变形成约9.1 nm 的小尺寸纳米粒子,高度均匀地分散在GO 表面,该催化剂的积炭层厚度大幅降低至2.3 nm。由此可以推断,铁物种与GO 中各种含氧官能团之间的相互作用对演变过程起关键作用[19]。

图6 新鲜和反应后催化剂的TEM 照片Figure 6 TEM images of the fresh and spent catalysts
反应后催化剂的穆斯堡尔谱数据如表2 所示。超精细磁场H 值为216、186 和112 KOe,表明χ-Fe5C2的存在[42,47,48]。H 值为169 KOe[49]和464 KOe[50]分别对应ε′-Fe2.2C 和Fe3O4。超顺磁性双重峰的IS(0.50 mm/s)和QS(1.07 mm/s)归属为结晶度较差的Fe(Ⅱ)或Fe(Ⅲ)氧化铁物种[51]。如表2所示,在340℃下反应50 h 后,纯Fe3O4逐渐还原碳化只形成了χ-Fe5C2,而Fe/GO 中的铁氧化物则逐渐还原碳化形成了高含量的ε′-Fe2.2C(44.8%)。在0.4 KFe/GO 中,K 的添加导致ε′-Fe2.2C 含量进一步增加,达到约58.9%。据文献报道,ε′-Fe2.2C 通常在高碳势(μc)环境中形成,粒径减小或助剂加入有利于稳定ε′-Fe2.2C[52,53]。因此,GO 加入带来的高碳势和碳化铁粒径的减少,可能是导致ε′-Fe2.2C含量增加的主要原因。此外,K 助剂有利于CO 解离增加μc,促进ε′-Fe2.2C 形成[54,55]。

表2 反应后催化剂的穆斯堡尔光谱Table 2 Mössbauer spectra of the spent catalysts
为了进一步确定反应后催化剂中铁物相组成,对催化剂进行了XRD 表征,结果如图7 所示。反应后的Fe3O4呈现χ-Fe5C2衍射峰[56],而Fe/GO中χ-Fe5C2衍射峰最强。由于χ-Fe5C2良好的结晶性导致不稳定的结晶性较差的ε′-Fe2.2C 很难在XRD 衍射峰中观察到[57]。然而,对于0.4 KFe/GO 和0.8 KFe/GO 催化剂,XRD 谱图中可以明显观察到ε′-Fe2.2C,这与穆斯堡尔谱的结果是一致的。
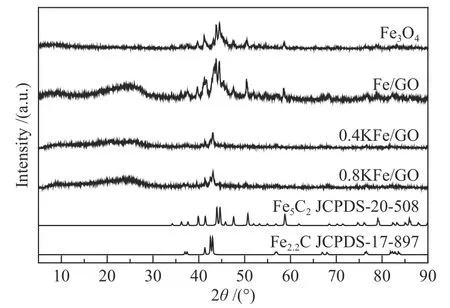
图7 反应后催化剂的XRD 谱图Figure 7 XRD patterns of the spent catalysts
2.3 催化性能
在340℃,1 MPa,H2/CO=1 和22.2 L/(gcat∙h)空速的条件下,评价了上述催化剂的FTS 反应性能,结果如图8、图9 和表3 所示。对于Fe3O4,反应最初5 h 催化剂快速失活,CO 转化率从40%急剧下降至8.5%。催化剂快速失活及高的甲烷选择性主要归因于Fe 的烧结,特别是积炭(图6(b))[17,30]。Fe/GO 不仅没有失活,相反,随反应的进行转化率逐渐增加,并最终达到高的稳定的催化活性(CO 转化率约84%)。结合图6(d)中的TEM 照片可以推测,这种反常的催化行为与Fe3O4微球的初始形态和结构演变密切相关。大尺寸的Fe3O4颗粒在反应条件下已经逐渐演变成小尺寸碳化铁纳米颗粒,从而导致活性位点数量大幅增加,且小尺寸纳米粒子有利于抑制催化剂积炭失活。此外,Fe/GO 显示了约31%的高的低碳烯烃()选择性,CH4选择性明显降低。由图4(b)的CO-TPD结果可知,GO 的加入有利于CO 的吸附和解离,从而提高催化剂表面C*物种浓度。确实,GO 添加的铁基费托催化剂的催化剂活性明显高于传统的共沉淀法制备的催化剂,可能主要源于其独特的电子性质。此外,由于制备方法导致GO 常常含有少量S[41]。研究表明,少量S 助剂的加入有利于低碳烯烃生成,同时降低CH4选择性[17]。几个催化剂费托反应的产物的碳数分布和α-烯烃含量如图9 所示。即便纯Fe3O4也显示较高的C2+α-烯烃选择性,可能源于铁基催化剂本质上具有比钴催化剂更弱的加氢能力,铁基催化剂更易于发生β-H 消除终止碳链增长,生成烯烃产物[52]。高的CH4选择性主要归因于沉积炭抑制了碳链增长,从而使物种转向发生连续加氢反应。相反,大幅降低的积炭速率导致Fe/GO 显示出低的CH4选择性,且α-烯烃在总烃和C2+烃中的选择性均很高,分别为80%和86%(图9(b)和表3)。

表3 不同催化剂的FTS 反应结果。Table 3 Catalytic performance of the FTS reactions over various catalysts
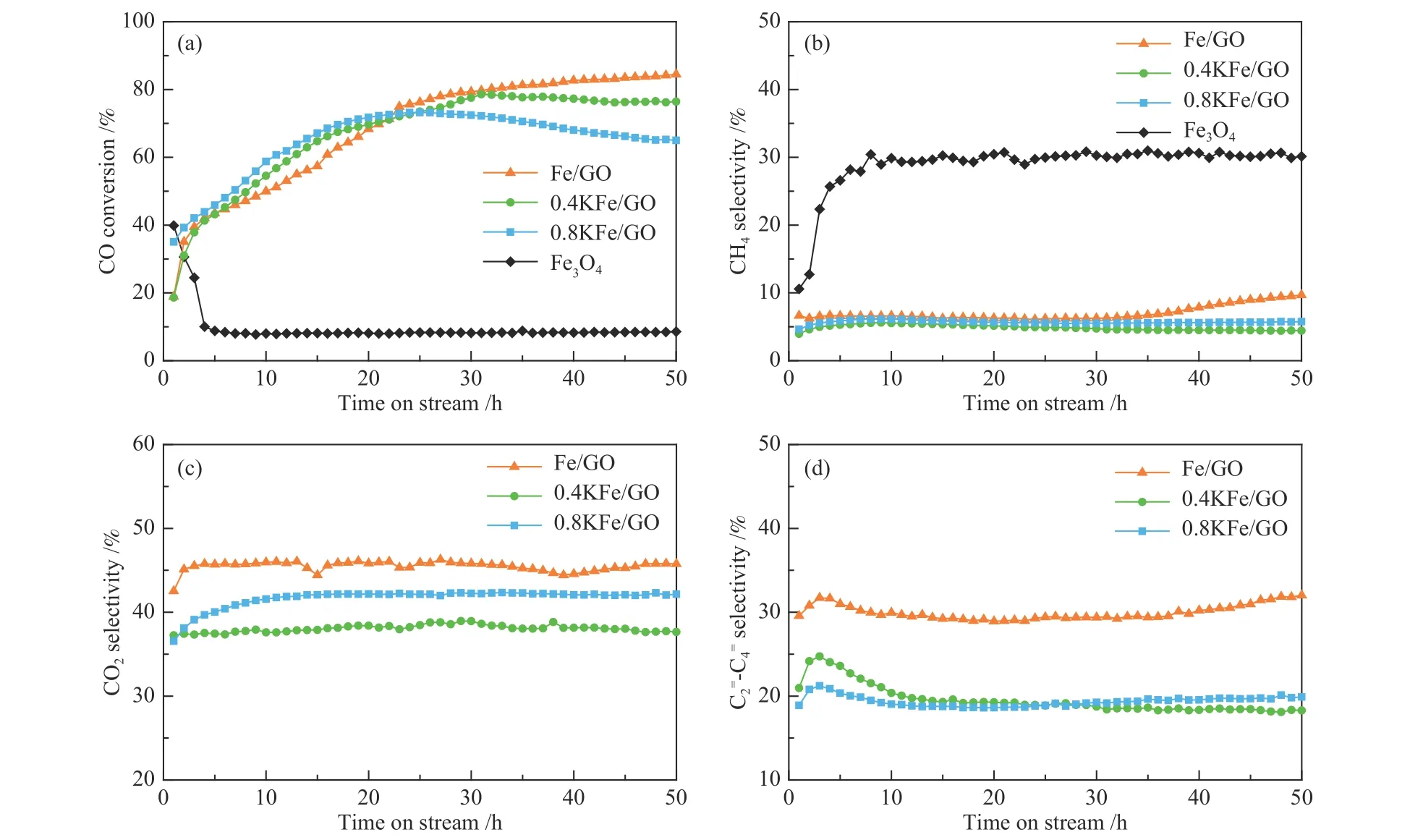
图8 Fe3O4 和Fe/GO 的催化性能Figure 8 Catalytic performances of Fe3O4 and Fe/GO catalysts

图9 反应的碳数分布Figure 9 Carbon number distribution (catalysts and reaction conditions are same as shown in Figure 8)
对于0.8KFe/GO 而言,当加入K 的量较多时,反应30 h 后出现明显失活,可能是由于过高K 含量显著提升CO 解离,从而导致积炭量增加。由图4(b)的CO-TPD 结果可知,K 的添加确实促进了CO 解离,导致CO 快速分解为C*物种[58,59]。由于随后的加氢和C−C 偶联等反应不能及时消耗催化剂表面的C*物种,导致活性位点被覆盖进而使催化剂失活。此外,K 的添加对产物分布也有较大影响。与Fe/GO 相比较,CH4和C2−4烃选择性降低,C5+选择性明显增加,表明K 抑制了CH4的形成并提升了链增长能力,链增长可能性增加源于K 提升了CO 解离导致催化剂表面更高的C*/H*比[13]。图5(a)中H2-TPR 结果表明,K 的添加抑制了H2的解离,从而有利于降低CH4选择性。此外,随着K 添加量的增加,CO2选择性出现先降低后升高的趋势。CO2选择性的降低可能与0.4KFe/GO 中高ε′-Fe2.2C 含量有关(如表2 所示)。众所周知,费托合成反应中的Boudouard 反应(CO →C*+O*;O*+CO → CO2)和WGS 反应(CO*+H2O→ CO2+H2)均能生成CO2[60]。作者的研究表明,Boudouard 反应在χ-Fe5C2上生成CO2更容易进行,因为与O*物种加氢生成H2O 相比,CO 与O*反应具有更低的能量壁垒[61]。文献报道,ε′-Fe2.2C 没有Boudouard 反应活性,具有低的CO2生成能力,而氧化铁是WGS 反应的活性相[49]。在本研究中,随着K 含量增加,氧化铁从16.1%逐渐增加到21.7%,ε′-Fe2.2C 略有下降,同时χ-Fe5C2含量相应增加,物相变化是导致CO2选择性增加的主要原因。
图10 和图11 给出了0.4KFe/GO 上H2/CO 比对催化活性和产物选择性的影响。随着H2/CO 比增加,催化剂的诱导期明显缩短,并达到了更高更稳定的催化活性(图10(a))。这种现象可归因于两方面的原因,一方面,H2分压提高有利于Fe3O4还原和随后的碳化[60];另一方面,CO 分压的降低和高的H2分压有利于避免CO 解离导致的炭沉积失活。此外,H2/CO 比的提高有利于减少CO2的形成,其选择性从38.3%降至31.6%,分别对应H2/CO比为1 和3 两种情况(图10(c)和表3)。H2/CO 比的提高使得表面H*浓度提高,有助于将CO*离解的O*物种加氢生成H2O,从而降低其与CO*反应生成CO2。此外,H*物种浓度的提高有利于CO 的H 辅助解离,减小直接解离形成O*后经Boudouard 反应生成CO2的可能性。从图10(d)和表3 可以看出,随着H2/CO 比从1 增加到3,选择性从18.5%增加到30.6%,这是由于高的H2/CO 比使催化剂表面C*物种浓度降低,使链增长可能性降低。如图11 和表3 所示,高H2/CO比没有明显改变C2+烃中的α-烯烃含量(86.1%),这与Co 基催化剂完全不同。对于Co 基费托催化剂,提高H2/CO 比将明显增加饱和烷烃选择性[62]。该结果表明铁基催化剂本质上更有利于α-烯烃的生成[57]。

图10 H2/CO 比对0.4KFe/GO 催化FTS 反应的影响Figure 10 Effects of the H2/CO ratio on FTS over 0.4KFe/GO catalyst
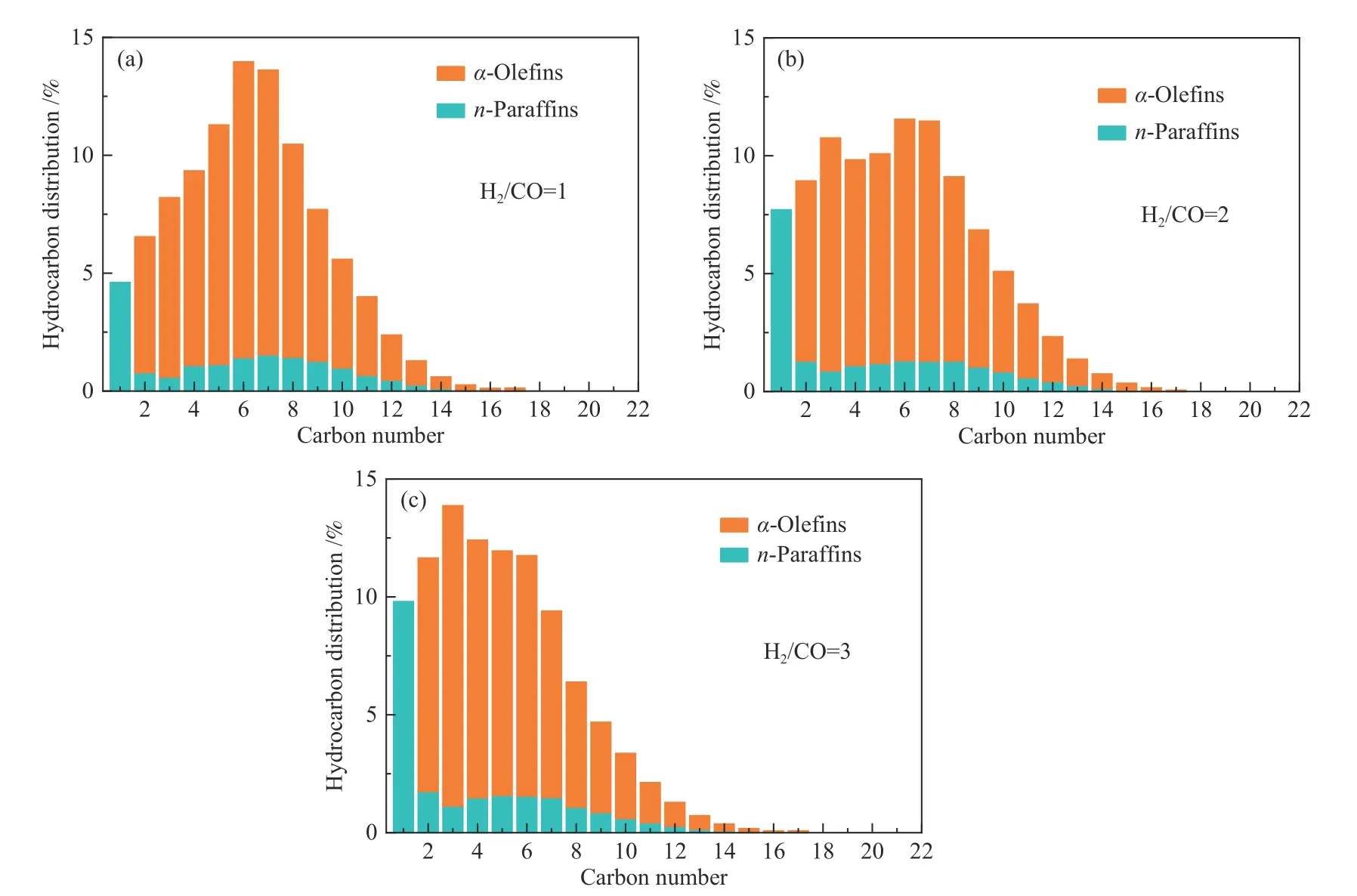
图11 反应的碳数分布(催化剂和反应条件同图10)Figure 11 Carbon number distribution (catalysts and reaction conditions are same as shown in Figure 10)
3 结 论
Fe/GO 催化剂在FTS 反应条件下逐渐演变成高度分散的小尺寸碳化铁纳米胶囊颗粒,表现出优异的活性、稳定性和α-烯烃选择性。K 的添加使Fe3O4在反应过程中演变生成更多的ε′-Fe2.2C相,有利于减小CO2选择性。增加H2/CO 比能在保证高的α-烯烃前提下进一步减少CO2选择性,同时减少炭沉积提高催化剂稳定性。
猜你喜欢
应用化工(2022年9期)2022-10-24
炼油与化工(2022年4期)2022-10-10
纺织标准与质量(2022年3期)2022-08-10
纺织标准与质量(2022年2期)2022-07-12
分析测试技术与仪器(2022年2期)2022-07-08
科学家(2022年4期)2022-05-10
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
当代化工(2020年2期)2020-03-18
分析化学(2017年12期)2017-12-25
山东农药信息(2013年6期)2013-08-09
