
砷/氢/氧体系中均相反应机理的研究
2021-08-15 07:51熊中朴陈文洋
燃料化学学报 2021年7期
熊中朴,于 洋,陈文洋,陈 娟
(山东大学 能源与动力工程学院,山东 济南 250061)
煤炭是中国重要的能源,而煤中含有的砷也是中国砷污染重要来源之一[1]。据估算,早在2005年中国燃煤排砷量已达到1564.4 t[2]。砷在人体内长期积累会造成砷中毒,并诱发皮肤、神经系统以及心血管疾病,近年来,中国也发生过多次因燃煤造成的砷中毒事件[3]。同时,煤直接燃烧产生的砷的氧化物也会使锅炉内的催化还原脱硝(SCR)催化剂中毒,进而造成氮污染[4]。随着人们环保意识的提高,砷等痕量元素形成的污染物也备受关注。在煤燃烧过程中,煤裂解形成的砷以气态形式在烟气中存在,受到氧气和水的氧化作用形成化合物[5]。由于砷各转化路径的反应速率不同,使生成的各价态的砷化物浓度不同。相比于单质砷,砷的化合物毒性更强,砷及其化合物毒性由强到弱排序为:AsH3>As3+>As5+>RAsX(有机砷化物)>As0[6]。因此,为了针对性地控制砷化物对环境的污染,分析各种砷化物转化的反应机理和速率尤为重要。
量子化学是计算化学中一种相对成熟的分析微观机理的手段,通过量子化学的计算可以确定反应路径上的各驻点的几何构型以及能量,并在该过程中计算出分子动力学和热力学参数,为砷的反应机理研究提供了理论依据[7]。近年来,很多学者采用量子化学的方法计算了燃煤过程中各种污染物的反应机理[8−13]。由于砷在燃烧过程中被氧气和水氧化而使形态发生转变,所以在研究砷转化微观机理应在含氢体系内进行,但目前,国内外的研究很少涉及砷/氢/氧体系燃烧反应微观机理研究。邹潺等[14]利用Gaussian09 软件包研究了砷与N2O、NO2以及NO 的反应机理,Urban 等[15]基于密度泛函理论研究了As+HCl=AsCl+H 反应动力学,Monahan 等[16]研究了砷与常见的自由基的反应机理,闫傲等[17]应用密度泛函理论研究了燃煤烟气中As 及AsO 和O2均相反应生成As2O3反应机理。但这些反应数量较少,不能完整地描述砷在燃烧时的转化过程,因此,对于砷在高温燃烧过程中的反应机理目前并没有完整的体系。
本研究利用Gaussian09 和GaussView 软件包通过微观机理研究,精确的测定砷燃烧过程发生的基元反应所需的活化能,拟合曲线后得到各基元反应的反应速率参数,为建立砷燃烧动力学模型奠定基础。
1 计算方法
本研究采用的计算方法为B3LYP,采用6-311G(3df,3pd)基组。在该理论水平下对有相关数据的标准物质O2、AsO 以及两种As2O3的主要构型(D3H 和GAUCHE)进行几何优化[17]。通过Gaussian09 软件包计算得到它们的几何构型如图1所示。

图1 优化后的AsO、O2、As2O3(D3H)和As2O3(GAMCHE)Figure 1 Optimized AsO,O2,As2O3 (D3H) and As2O3 (GAMCHE)
从表1 中可知,AsO 和As2O3(D3H)的构型计算结果与实验值基本符合,而O2和As2O3(GAMCHE)的计算结果与实验值相比存在一定误差,但误差在可接受范围内。以上数据表明,在该理论水平下进行砷/氢/氧体系反应计算所得到的结果可信度较高。

表1 计算值与文献值对比Table 1 Comparison of calculated values and literature values
考虑到砷元素形成的化合物种类较多,微观反应机理较复杂,且氧/氢之间的反应速率常数在美国国家标准与技术研究院(NIST)数据库较为齐全,因此,本文仅限于研究砷/氧/氢燃烧动力学模型中不含氧氢之间反应的13 个反应。
通过量子化学的方法,构建分子模型,本文采用GaussView 程序包构建分子模型,应用Gaussian09程序包对构型进行优化。在B3LYP/6-311G(3df,3dp)理论水平下对砷/氢/氧体系中的反应进行研究。在研究过程中先对反应物和产物进行优化,而后猜测并搜索该反应的过渡态(TS)。确认反应物和生成物全为实频以及搜索得到的过渡态有且仅有一个虚频并确认振动方向与反应方向相同后,再由过渡态搜索内禀反应坐标(IRC),所得到的IRC 连接反应物和生成物并验证反应路径,进一步证明搜索得到的过渡态为该反应真正的过渡态。最后通过KiSThelP 拟合得到阿伦尼乌斯三参量修正方程中的参数。考虑到烟气温度从炉膛出口流经烟道排出过程中温度不断降低,因此,本文拟合计算温度500−2000 K 下的平均参数。
2 结果与讨论
2.1 反应分析
经过Gaussian09 程序包的过渡态搜索,共得到13 个砷/氢/氧体系在燃烧过程中发生的基元反应,如表2 所示。

表2 砷参与的基元反应Table 2 Elementary reactions of Arsenic
2.2 反应机理
本研究以AsO2OH+H2O=H3AsO4反应为例,对砷微观反应机理进行介绍。首先对反应物单体进行优化,基本确定反应物单体构型。再对反应的一阶鞍点初猜构型进行优化,即可得到该反应的过渡态。在采用Gaussian09 程序包搜索AsO2OH+H2O=H3AsO4的过渡态时,首先确定发生反应的位置,基于此改变反应物构型,并使之尽量与生成物接近,而后不断对构型进行微调,进而搜索到过渡态。经过Gaussian09 软件包搜索得到中间体、过渡态以及生成物构型见图2。反应AsO2OH+H2O=H3AsO4在B3LYP/6-311G(3df,3dp)的理论水平下的反应过程如图3 所示。

图2 AsO2OH+H2O=H3AsO4 反应物、过渡态以及生成物构型Figure 2 Geometries of intermediate,transition state and product of AsO2OH+H2O=H3AsO4

图3 AsO2OH+H2O=H3AsO4 反应过程示意图Figure 3 Reactant process analysis of AsO2OH+H2O=H3AsO4
表3 中列出了反应物、过渡态和生成物的振动频率。显然,该反应的过渡态只有一个虚频,虚频大小为−1358.59 cm−1。虚频中原子振动方向为7H 由6O 向4O 方向振动,振动方向与反应方向相同,表明该过渡态可信度较高,为进一步确认过渡态的正确性,利用内禀反应坐标确认该反应路径。

表3 AsO2OH+H2O=H3AsO4 反应各稳定点振动频率Table 3 Frequencies of stable points of AsO2OH+H2O=H3AsO4
图4 为内禀反应坐标图,由搜索到的过渡态连接上了反应物和生成物并确认了AsO2OH+H2O=H3AsO4为基元反应。在反应过程中,水分子中的氢原子与羟基之间的键逐渐增长,由0.96037 Å 增长到 1.17149 Å 最后达到∞,砷原子与其中一个氧原子的π 键断裂。羟基靠近砷原子形成砷氧键(∞−2.00147 Å −1.74838Å),氢原子靠近有孤电子的氧原子形成氢氧键(∞−1.35277 Å −0.96486 Å),形成H3AsO4。
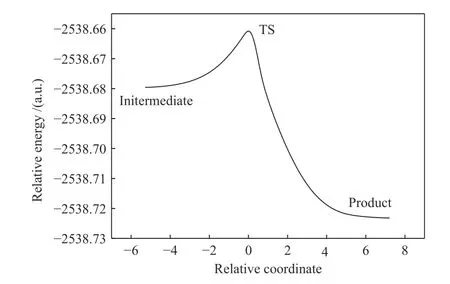
图4 AsO2OH+H2O=H3AsO4 内禀反应坐标Figure 4 Intrinsic reaction coordinate diagram of AsO2OH+H2O=H3AsO4
由IRC 图可知,该反应的正向反应活化能较小、逆向反应活化能较大,证明该反应更容易正向进行。结合零点校正,在298 K 下各物质能量表以及反应过程能量变化如表4 和图5 所示。

表4 AsO2OH+H2O=H3AsO4 反应过程能量变化Table 4 Energy change in AsO2OH+H2O=H3AsO4 reaction process

图5 AsO2OH+H2O=H3AsO4 反应过程能量变化Figure 5 Gibbs energy change in AsO2OH+H2O=H3AsO4 reaction process
2.3 反应常数的计算
优化后的构型经过校正后得到相对能量,便可用于计算反应的活化能。本文所研究的温度范围较宽,因此,应拟合阿伦尼乌斯三参量修正方程。
本研究采用KiSThelP 软件包拟合求得反应动力学参数。KiSThelP 作为一款以Java 为基础环境可以读取Gaussian 频率输出文件的程序,基于过渡态理论反应模型计算反应速率常数k,并在虚频的基础上做Wigner 隧道校正,拟合阿伦尼乌斯三参量修正方程,得到反应的的动力学参数[18]。拟合曲线如图6。

图6 KiSThelP 拟合阿伦尼乌斯曲线Figure 6 KiSThelP fitting Arrhenius curve
AsO2OH+H2O=H3AsO4过程中先由弱作用力形成的稳定的复合物,再跨越能垒生成产物,本研究将该基元反应以单分子反应形式进行处理。该过程的反应路径简并度为1;由于该计算在B3LYP/6-311G(3df,3pd)理论水平下计算,参考B3LYP/6-31G(2df,2p),谐振频率校正因子设置为0.98[19,20]。拟合结果为:A=9.80×109,n=0.52,Ea=7982 cal/mol。
根据上述计算,该反应的反应速率在温度范围内受温度影响较小但k 值较大,反应进行程度较大,同时也可以证实生成的AsO2OH 会迅速发生转化。
其他12 个基元反应采用同样的的处理方法并经过KiSThelP 拟合,计算得到各反应动力学参数如表5 所示。

表5 砷参与的各基元反应反应动力学参数Table 5 Kinetic parameters of each elementary reaction of Arsenic
3 结 论
本研究利用Gaussian09 和GaussView 软件包在B3LYP/6-311G(3df,3pd)的水平对砷反应的微观机理进行探究,优化了路径上各驻点的几何构型并计算能量。最终确定了13 个由砷参与的基元反应。最后根据过渡态理论利用KiSThelP 程序包计算了这13 个反应的动力学参数,为后续研究砷/氢/氧体系燃烧动力学打下了基础。
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
兵工学报(2022年2期)2022-05-22
西北工业大学学报(2022年1期)2022-04-22
兵工学报(2021年4期)2021-06-19
天津理工大学学报(2021年2期)2021-06-03
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
兵工学报(2020年12期)2020-02-06
科学导报(2018年30期)2018-05-14
中学化学(2017年5期)2017-07-07
中学化学(2016年4期)2016-05-30
