
禽致病性大肠杆菌clpV基因缺失株的构建及其对Ⅰ型菌毛主要基因表达的影响
2021-08-10 07:27钟昊然王培莉陈艳飞朱国强李建基崔璐莹董俊升
中国兽医学报 2021年6期
钟昊然,王培莉,陈艳飞,孟 霞,朱国强,李建基,崔璐莹,董俊升,王 亨*
(1.扬州大学 兽医学院,江苏省动物重要疫病与人兽共患病防控协同创新中心,江苏 扬州 225009;2.教育部农业与农产品安全国际合作联合实验室,江苏 扬州 225009)
禽致病性大肠杆菌(avian pathogenicEscherichiacoli,APEC)是我国禽大肠杆菌病的主要感染病原之一,至今尚无科学有效的防治措施[1]。APEC与新生儿脑膜炎大肠杆菌(neonatal meningitisE.coli,NMEC)的毒力基因存在高度同源性,且已有APEC TW-XM菌株能引起禽脑膜炎的报道[2-3]。随着集约化养殖的发展,APEC已成为一种潜在的人兽共患病原菌,有使婴儿感染新生儿脑膜炎的潜在风险,具有重要的公共卫生意义[4]。大肠杆菌作为革兰阴性菌,可释放一种多功能杀伤性武器T6SS(type Ⅵ secretion system,T6SS),它能通过分泌效应蛋白作为武器攻击靶细胞从而参与致病过程[5]。ClpV是一种拆解T6SS的元件,可允许细胞在ATP的驱动下完成T6SS的回收和组装[6]。研究证实ClpV在假单胞菌(Pseudomonasplecoglossicida)[7]的致病性方面有着重要作用,但ClpV在APEC脑膜炎的致病过程中发挥何种功能却鲜有研究。
Ⅰ型菌毛是众多革兰阴性致病菌的表面附属结构,是主要的黏附因子,在细菌侵入机体的过程中扮演着重要角色[8-9]。已有研究表明,在E.coliK1所引起的新生儿脑膜炎中,Ⅰ型菌毛的黏附起决定性作用[10]。虽然T6SS与Ⅰ型菌毛均参与新生儿脑膜炎的致病过程,但两者之间的联系还有待进一步诠释。本研究利用Red同源重组系统,成功构建了TW-XM菌株T6SS中的clpV基因缺失株与回补株,进一步探究了clpV基因对Ⅰ型菌毛合成的影响,为后续研究APEC致脑膜炎的机理奠定了基础。
1 材料与方法
1.1 菌株与质粒菌株APEC TW-XM、DH5α以及质粒pKD46、pKD3、pCP20均由扬州大学朱国强教授馈赠。
1.2 培养基与主要试剂、仪器氯化钠、胰蛋白胨、酵母提取物购自英国Oxoid公司;氨苄青霉素、氯霉素购自上海生工生物工程有限公司;Ex Taq DNA聚合酶、琼脂糖凝胶DNA回收试剂盒、DL2000 DNA Marker、DL5000 DNA Marker均购自日本TaKaRa公司;L-阿拉伯糖购自美国Sigma公司;TRIzol购自北京天根生化科技有限公司;ChamQ SYBR qPCR Master Mix、HiScript®两步法RT-qPCR系列试剂盒均购自南京诺唯赞生物科技股份有限公司;MicroPulser Electroporator、CFX Connect荧光定量PCR仪均购自美国Bio-Rad公司;NanoDrop 2000核酸测定仪购自美国Thermo公司。
1.3 引物设计与合成根据GenBank APEC TW-XM菌株(NO.KF678349.1)中已知的clpV基因序列设计用于扩增clpV基因的引物,命名为clpV-F/clpV-R,并且clpV-F/clpV-R可用于鉴定clpV基因缺失株与回补株的构建是否成功。此外,设计1对5′端与clpV基因两翼同源(用下划线标出),3′端与pKD3中间的氯霉素抗性基因cat两侧序列同源的重组引物,命名为ΔclpV-Cm-F/ΔclpV-Cm-R。回补引物pBR-clpV-F/pBR-clpV-R则根据无缝克隆技术设计[11]。所有引物均由南京擎科生物技术公司合成,引物序列见表1。

表1 构建clpV基因缺失株与回补株的引物序列
1.4 一次同源重组菌的构建与鉴定按照丁雪燕等[12]的方法,将clpV线性片段进行扩增、回收与纯化。同时,将含有pKD46的TW-XM接种于含有L-阿拉伯糖(30 mmol/L)的LB液体培养基中,使pKD46中的Exo、Bet及Gam蛋白充分表达,制备成感受态细胞。随后将先前制备的clpV线性片段和TW-XM感受态细胞的电击转化产物涂布于含有Amp和Cm抗性的LB平板,筛选阳性转化子,以clpV-F/clpV-R为引物进行PCR鉴定。PCR反应体系为Ex Taq DNA聚合酶12.5 μL,引物clpV-F/clpV-R各1.0 μL,模板1.0 μL,补ddH2O至25.0 μL。PCR程序为94℃ 4 min;94℃ 30 s,57℃ 30 s,72℃ 30 s,25个循环;72℃延伸10 min。鉴定正确后由南京擎科生物技术公司测序,并命名为TW-XM△clpV::cat。
1.5 二次同源重组菌的构建与鉴定将TW-XM△clpV::cat制备成感受态细胞,并将pCP20质粒导入感受态细胞中,随后将转化产物涂于含Amp和Cm双抗性的LB平板上,筛选对Amp和Cm均敏感的阳性转化子。以clpV-F/clpV-R为引物进行PCR鉴定,反应体系与反应程序同1.4。鉴定正确后由南京擎科生物技术公司测序,并命名为TW-XM△clpV。
1.6clpV基因回补株的构建与鉴定以pBR-clpV-F/pBR-clpV-R为引物,TW-XM为模板,PCR扩增得到clpV基因,反应体系为Q5 High Fidelity 2×Master Mix 25.0 μL、pBR-clpV-F/pBR-clpV-R各2.5 μL、模板1.0 μL、补ddH2O至50.0 μL。PCR程序为95℃预变性 5 min;95℃ 30 s,57℃ 30 s,72℃ 60 s,30个循环;最后72℃延伸10 min。经测序验证正确后克隆至表达载体pBR322中,随后将重组质粒pBR322-clpV转化至TW-XM△clpV,得到回补株TW-XMC△clpV,并经PCR和测序鉴定,PCR反应体系与反应程序同1.4。
1.7clpV基因缺失株与回补株的遗传稳定性试验将缺失株TW-XM△clpV与回补株TW-XMC△clpV在LB平板上连续传代,取第1,10,20,30代的单菌落制备模板,进行PCR鉴定以分析各菌株遗传稳定性,PCR反应体系与反应程序同1.4。
1.8 细菌总RNA的提取挑取野生株TW-XM、缺失株TW-XM△clpV与回补株TW-XMC△clpV接种于LB液体培养基中。培养至D630为1时,用TRIzol法提取细菌总RNA[13]。随后采用NanoDrop 2000核酸测定仪测定总RNA浓度,总RNA经HiScript®两步法RT-qPCR系列试剂盒反转录得到cDNA。
1.9 荧光定量PCR检测Ⅰ型菌毛主要基因的表达自NCBI GenBank 获得大肠杆菌Ⅰ型菌毛主要基因(fimA、fimB、fimC、fimD、fimE、fimF、fimG、fimH、fimI)及内参基因gapA的序列,设计引物交至南京擎科生物技术公司合成,引物序列见表2。使用ChamQ SYBR qPCR Master Mix进行荧光定量RT-qPCR。反应体系为5 μL ChamQ SYBR qPCR Master Mix,1 μL 正向引物,1 μL 反向引物,2 μL cDNA 模板,补去离子水至10 μL。反应程序为95℃预变性2 min;95℃ 10 s,58℃ 30 s,40个循环,95℃ 15 s,60℃ 5 s;95℃ 50 s。数据分析采用相对定量方法(2-△△Ct)。

表2 荧光定量PCR引物信息
1.10 统计学分析数据处理采用SPSS 17.0软件,使用单因素方差分析(One-way ANOVA),*或#表示差异显著(P<0.05),**或##表示差异极显著(P<0.01)。
2 结果
2.1 融合PCR产物的鉴定如图1所示,PCR扩增出两侧与clpV基因上、下游序列同源、中间为cat基因的融合PCR产物,大小约为1 100 bp,与预期一致。表明扩增出含clpV基因同源臂的cat基因序列。

M.DL2000 DNA Marker;1.融合PCR产物
2.2 同源重组菌的鉴定如图2所示,PCR结果显示:野生菌clpV扩增片段约2 200 bp,TW-XM△clpV::cat重组体约1 800 bp,表明一次同源重组菌构建成功。如图3所示,通过电转化将pCP20导入TW-XM△clpV::cat,经二次重组后筛选阳性克隆,PCR鉴定显示野生菌clpV扩增片段约2 200 bp,重组体约为700 bp。表明二次同源重组菌构建正确。进一步对上述筛选的突变株clpV基因两侧序列克隆和序列测定。结果显示经二次重组后,突变株中的cat基因已完全消除,表明成功构建clpV基因突变株TW-XM△clpV。

M.DL5000 DNA Marker;1.野生株clpV基因;2.含氯霉素抗性基因cat的clpV一次重组基因

M.DL5000 DNA Marker;1.野生株clpV基因;2.clpV基因二次重组后片段
2.3clpV基因回补株的鉴定如图4所示,PCR检测结果显示2个特异性条带,其中700 bp左右条带为clpV缺失株的二次同源重组条带,2 200 bp左右条带为回补质粒pBR322-clpV中的clpV基因片段。测序验证显示结果正确,表明回补株TW-XMC△clpV构建成功。
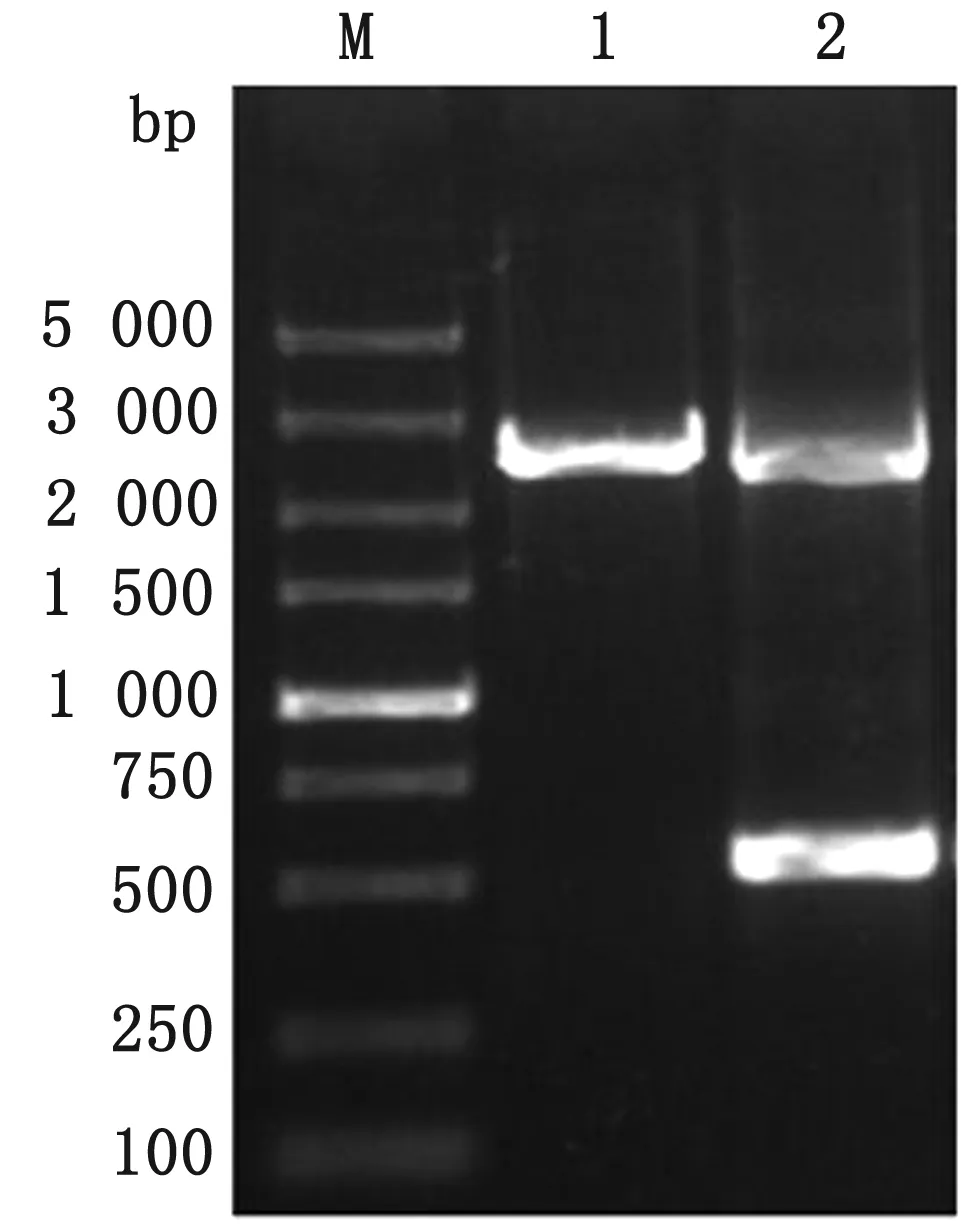
M.DL5000 DNA Marker;1.野生株clpV基因;2.clpV基因回补片段
2.4clpV基因缺失株与回补株的遗传稳定性测定如图5所示,缺失株TW-XM△clpV与回补株TW-XMC△clpV经连续传30代后,对不同代数的缺失株与回补株分别进行PCR鉴定。结果显示,不同代数的缺失株均可扩增出二次同源重组的特异性条带,而不同代数的回补株均可扩增出二次同源重组的特异性条带以及回补质粒pBR322-clpV携带的clpV基因片段。各菌株均未发生突变,表明缺失株TW-XM△clpV与回补株TW-XMC△clpV具有良好的遗传稳定性。
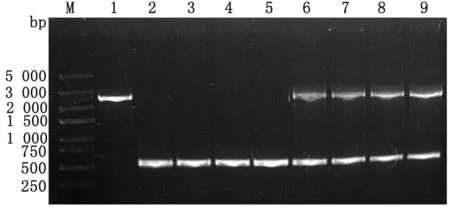
M.DL5000 DNA Marker;1.野生株clpV基因;2~5.缺失株TW-XM△clpV第1,10,20,30代中的二次重组片段;6~9.回补株TW-XMC△clpV第1,10,20,30代中的clpV基因回补片段
2.5clpV基因缺失对Ⅰ型菌毛主要基因mRNA表达的影响如图6所示,缺失株TW-XM△clpV与野生株APEC TW-XM相比,fimA、fimB、fimC、fi-mD、fimE、fimF、fimG、fimH、fimI基因的mRNA相对表达量均呈下降趋势。其中fimC、fimF、fimG、fimI的mRNA相对表达量显著下降(P<0.05);fimB、fimD、fimE的mRNA相对表达量则极显著下降(P<0.01);TW-XM△clpV组与APEC TW-XM组相比fimA与fimH的mRNA相对表达量则无统计学意义;回补株TW-XMC△clpV的各基因表达量得到了不同程度的恢复。

注:*或**表示与野生株APEC TW-XM相比,差异显著(P<0.05)或极显著(P<0.01)
3 讨论
基因敲除技术是一种将基因同源重组技术和胚胎干细胞技术相结合的新型分子生物学技术。Red同源重组是通过自身RecA系统中的RecA和RecBCD蛋白,使2个DNA分子之间交换片段以达到序列敲除的目的。1998年,ZHANG等[14]发现大肠埃希菌属中存在具有重组功能的RedE/RecT系统,而Red同源重组中的Exo和Beta蛋白也具有相同的功能。因此Red同源重组被认为是适合大肠杆菌的一种基因敲除方法[15]。
2006年,PUKATZKI等[16]在霍乱弧菌中发现了T6SS,此后对于T6SS结构、功能机制的研究便不断深入。现如今T6SS被认为是许多革兰阴性致病菌的潜在毒力决定因子,它能够促进细菌病原体之间,以及细菌病原体和宿主之间的相互作用,并在细菌将蛋白运输到胞外的过程中发挥重要作用[17-18]。T6SS的核心组分之一ClpV是AAA+ATP超家族的成员,可允许细胞在ATP驱动下完成T6SS组分的回收和组装,并为T6SS分泌效应蛋白提供能量[5]。
先前已有学者利用Red同源重组技术成功敲除了APEC CE129菌株中T6SS组分的hcp和APEC DE719菌株中T6SS组分的evfC基因,并对相应的缺失株进行生物学特性分析,结果显示以上基因的缺失会对APEC的运动性、生物被膜形成能力、黏附侵袭能力、胞内存活能力以及对雏鸭的致病力产生影响[12,19-21]。证明T6SS参与了APEC的致病过程。TANG等[7]则利用RNA干扰技术使假单胞菌(Pseudomonasplecoglossicida)中的clpV基因沉默,进而在假单胞菌与大黄鱼(Larimichthyscrocea)的转录组测序结果中发现clpV基因的沉默可下调Toll样受体信号通路、MAPK信号通路以及细菌鞭毛合成相关基因的表达。证明clpV基因在假单胞菌的致病过程中也发挥重要作用。本试验通过Red同源重组技术构建了APEC TW-XM菌株中T6SS的clpV基因缺失突变株,为后续研究clpV对APEC TW-XM的致病性影响奠定基础。
Ⅰ型菌毛是大肠杆菌的重要毒力因子,它可以通过促进细菌黏附、侵袭到宿主黏膜表面、引发机体的免疫应答等途径增强细菌的致病力[9,23]。MA等[23]和FERNANDA等[9]在分别敲除了APEC TW-XM和APEC SEPT362中T6SS的hcp基因后发现细菌对小鼠脑微血管内皮细胞(b.End3)和HeLa细胞的黏附能力均下降,从而导致菌株的致病力降低。以上研究证实T6SS与细菌的黏附存在一定的联系,但具体机制还需进一步研究。
KISIELA等[24]发现大肠杆菌Ⅰ型菌毛的表达至少需要9个基因(fimA、fimB、fimC、fimD、fimE、fimF、fimG、fimH、fimI)。本试验在成功构建clpV基因缺失株与回补株后,对以上9个基因的mRNA相对表达量进行检测。结果显示与野生株相比,在缺失clpV基因后,各基因的mRNA相对表达量均呈下降趋势,其中fimC、fimF、fimG、fimI的mRNA相对表达量显著下降;fimB、fimD、fimE的mRNA相对表达量则极显著下降。说明敲除T6SS核心组分的clpV基因会下调Ⅰ型菌毛的表达,进而有可能对细菌的黏附侵袭能力产生影响。
本试验中clpV基因的成功敲除,有助于深入了解T6SS与Ⅰ型菌毛之间的作用机制,为研究APEC的致病机理奠定基础,并有可能为禽大肠杆菌病疫苗的研发和防治提供新的靶标。
猜你喜欢
华人时刊(2023年1期)2023-03-14
汉字汉语研究(2021年2期)2021-08-30
传染病信息(2021年6期)2021-02-12
汉字汉语研究(2019年2期)2019-08-27
现代检验医学杂志(2016年2期)2016-11-14
河北书画研究(2016年3期)2016-04-28
四川畜牧兽医(2016年1期)2016-04-05
右江医学(2014年1期)2014-03-22
云南畜牧兽医(2014年2期)2014-02-28
中国预防兽医学报(2013年10期)2013-09-10
