
磁性固相萃取结合GC-MS检测烟叶中拟除虫菊酯类农药残留
2021-08-06 09:37杨玉明刘妧晨刘智敏司晓喜刘宏程许志刚
分析测试学报 2021年7期
杨玉明,刘妧晨,刘智敏,李 娇,司晓喜,林 涛,刘宏程,许志刚*
(1.昆明理工大学 理学院,云南 昆明 650500;2.云南中烟工业有限责任公司技术中心,云南 昆明 650231;
3.云南省农业科学院 质量标准与检测技术研究所,云南 昆明 650223)
拟除虫菊酯是20世纪70年代人工合成的一类杀虫剂,因具有廉价、杀虫谱广、低毒性、低残留、对环境友好等特点[1],被广泛用于蔬菜、茶叶等农产品生产[2-3]。目前,拟除虫菊酯类杀虫剂已发展成为继有机磷类农药、氨基甲酸酯类农药后使用最广的第三大类杀虫剂[4-5]。然而,已有研究发现拟除虫菊酯类杀虫剂在自然环境下降解速率慢,对光与热具有一定的稳定性,环境残留的拟除虫菊酯类杀虫剂会引发毒副作用[6-7]。此外,环境中残留的拟除虫菊酯类杀虫剂会通过食物链富集进入机体,危害人等哺乳动物的心血管、免疫力和生殖系统等[8-9]。因此,拟除虫菊酯类杀虫剂的残留问题受到普遍重视,如欧盟、美国等限定烟草中菊酯类农药最高残留量为0.5 mg/kg[10];我国国家标准YC/T 405.2-2011也将拟除虫菊酯类杀虫剂残留量设定为烟草中的检测项目并严格规范[11],但仍有烟草中检出拟除虫菊酯类农药的报道[12-14]。
目前,拟除虫菊酯类杀虫剂残留的检测方法有高效液相色谱法[15]、毛细管气相色谱法[16]、液相色谱-串联质谱法[17]、气相色谱-串联质谱法[18]等。但烟草等农产品中存在拟除虫菊酯类杀虫剂含量低、基质复杂、分析物种类多以及化合物间结构与性质差异小等问题,分析前一般需进行固相萃取[19]、分散固相萃取[20]、分散液相微萃取[21]、磁固相萃取[22]等样品前处理过程。其中磁性固相萃取(Magetic solid phase extraction,MSPE)是由Šafaříková等[23]于1999年在固相萃取技术的基础上发展的一种高效样品前处理方法,因吸附剂具有磁性,可在外加磁场中实现目标分析物与基质的快速分离,前处理操作简单,且磁性吸附剂易于回收和重复利用。环糊精是一类天然环状低聚糖化合物,具有疏水内腔和亲水外壳,且外壳含有活性羟基,使得环糊精可与有机分子、无机分子等分析物发生吸附作用[24-25],已广泛用于化工、食品、医疗等领域。环糊精作为吸附剂还需一定基质,使其可重复利用与回收[26]。
本研究选择无机材料SiO2为吸附剂基质,采用水热反应法制备了磁性Fe3O4纳米粒子,涂覆SiO2后,通过烷基偶联试剂γ-缩水甘油醚氧丙基三甲氧基硅烷键合上β-环糊精,得到Fe3O4@SiO2@β-CD磁性固相萃取剂,结合气相色谱-质谱实现了烟草样品中4种拟除虫菊酯类杀虫剂残留的分析。
1 实验部分
1.1 仪器与试剂
Clarus 600 GC气相色谱仪、Clarus 600 MS质谱检测器(美国珀金埃尔默公司);FV-SK-02型涡旋振荡器(美国莱伯特公司);KQ-700V型超声波清洗机(昆山市超声仪器公司);水热反应釜(含聚四氟乙烯内胆,南京瑞尼克科技有限公司);NDO-400型定温恒温干燥箱(上海爱朗仪器有限公司);79HW-1电子调温模拟控温磁力加热搅拌器(江苏优卓诺仪器制造有限公司);BT125D型电子天平仪器(德国赛多利斯科学仪器有限公司);Nova NanoSem 450扫描电镜(美国FEI公司);TENSOR27红外光谱仪(德国Bruker公司);PANayltical Empyrean型X射线衍射仪(PANalytical公司);AUTOSORBIQ-MP微孔吸附材料分析测试仪(美国康塔仪器公司);0.22μm尼龙有机系滤膜(上海新亚净化器件厂)。
FeCl3·6H2O(98%)、乙二醇(AR,98%)、醋酸钠(AR,99%)、聚乙二醇(PEG-2000)(Average Mn2000)、γ-缩水甘油醚氧丙基三甲氧基硅烷(KH560,98%)均购于阿拉丁公司;甲醇、乙腈等溶剂购于四川西陇化工有限公司;色谱级甲醇和乙腈等溶剂购于天津市津东天正精细化学试剂厂;NH3·H2O(25%)和N,N-二甲基酰胺(DMF)购于天津市风船化学试剂科技有限公司;工业氮气购于昆明融泰工贸有限公司;纯净水购于杭州娃哈哈集团有限公司;丙酮购于重庆川东化工(集团)有限公司;盐酸(36%~38%)购于云南杨林工业开发区汕滇药业有限公司;乙醇(分析纯)购自天津市致远化学试剂有限公司;氢化钠(NaH,80%)购自山东西亚化学股份有限公司;α-CD、β-CD、γ-CD购于山东滨州智源生物有限公司;联苯菊酯、甲氰菊酯、氟氯氰菊酯、溴氰菊酯4种标准品纯度大于95%均购于阿拉丁公司;烟草叶片采自云南红河州当地。
1.2 环糊精磁性复合材料的制备
准确称取2.7 g FeCl3·6H2O溶于50 mL乙二醇(EG)中,室温剧烈搅拌30 min后,再加入7.2 g醋酸钠和2.0 g聚乙二醇(PEG-2000),继续搅拌1 h,然后将溶液转移至含有聚四氟乙烯衬里的不锈钢高压釜中,密封后于200℃下加热8 h,将溶液中生成的黑色沉淀物冷却至室温并依次用乙醇和超纯水洗涤数次直至溶液pH值呈中性,得到的Fe3O4粉末于60℃下真空干燥。
准确称取1.0 g Fe3O4,加入10 mL 0.1 mol/L盐酸,超声处理10 min,通过磁分离除去液体,待固体物质上残留液体自然挥干,向有黑色颗粒的烧杯添加80 mL乙醇,再加入20 mL超纯水和2.5 mL NH3·H2O,搅拌均匀后,快速加入0.5 mL四乙氧基硅烷(TEOS)并在室温下搅拌12 h,得到Fe3O4@SiO2。
精确称取1.5 gβ-环糊精(使用前于90℃真空干燥8 h)溶于50 mL无水N,N-二甲基甲酰胺(DMF)中,加入0.15 g氢化钠并于室温下搅拌30 min,滤去未反应的固体物质,向滤液中加入0.5 gγ-缩水甘油醚氧丙基三甲氧基硅烷(KH560),氮气保护下于90℃反应5 h,再加入2.0 g Fe3O4@SiO2,提高温度至110~120℃继续反应24 h,将反应液冷却至室温,过滤,产物依次用DMF、二次蒸馏水、甲醇和丙酮洗涤4~5次,滤干后,将产物于100℃真空干燥24 h,得黑色固体Fe3O4@SiO2@β-CD,制备流程见图1。相同条件下制备Fe3O4@SiO2@α-CD和Fe3O4@SiO2@γ-CD用于萃取对照试验。
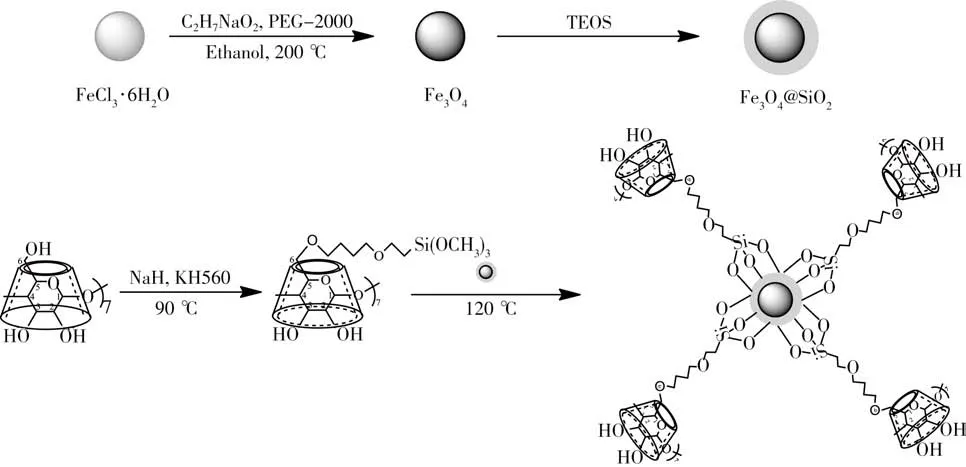
图1 β-环糊精磁性复合材料的制备流程图Fig.1 Diagram of preparation ofβ-cyclodextrin magnetic composite materials
1.3 样品前处理
烟草烟叶样品由云南中烟工业有限责任公司技术中心提供,3种烟叶样品经初烤后参考文献处理[27]:先将样品粉碎,准确称取100 mg于50 mL离心管中,加入2 mL甲醇,超声处理30 min,将目标分析物从烟草烟叶组织中提取到甲醇溶液中,超纯水定容至50 mL,并加入50 mg Fe3O4@SiO2@β-CD吸附剂,以150 r/min振荡提取120 min。磁分离弃上层溶液,待吸附材料中含有的少量液体自然挥干后加入2 mL甲醇(色谱纯),超声解吸5 min,再次磁分离得上层清液,过0.22μm尼龙有机相滤膜后,待GC-MS测定,每种样品平行测定3次。
1.4 色谱-质谱条件
DB-5 MS弹性石英毛细管色谱柱(30 m×0.25 mm i.d.×0.25μm d.f.,美国Agilent公司);进样口温度300℃,载气为氦气(纯度≥99.999%),恒流流速1.0 mL/min;进样量1μL,分流进样,分流比10∶1。程序升温:初始温度150℃,保持1 min,以10℃/min升至210℃,再以3℃/min升至300℃,保存5 min。电离方式:电子源轰击(EI);电子能量:70 eV;离子源温度:200℃;传输线温度:280℃;溶剂延迟:5.50 min;选择离子模式(SIM)检测。
2 结果与讨论
2.1 环糊精磁性复合材料的结构表征
2.1.1 电镜表征 采用扫描电镜对制备的Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD进行表征。结果显示:Fe3O4键合SiO2和β-CD后,表面结构和小球尺寸均发生了明显变化。结果显示:裸露的Fe3O4表面比较均匀且呈球形,平均直径约214.8 nm(图2A)。经SiO2修饰后,尺寸增大至227.6 nm得到Fe3O4@SiO2(图2B),再经β-CD修饰后尺寸为263.3 nm(图2C),修饰后的小球表面明显比Fe3O4更粗糙、多孔,由此表明SiO2和β-CD已键合在Fe3O4表面成功制备了Fe3O4@SiO2@β-CD复合材料。

图2 Fe3O4(A)、Fe3O4@SiO2(B)和Fe3O4@SiO2@β-CD(C)的扫描电镜图Fig.2 Scanning electron microscope pictures of Fe3O4(A),Fe3O4@SiO2(B)and Fe3O4@Si O2@β-CD(C)
2.1.2 红外光谱表征β-CD、Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD的红外光谱图见图3。由β-CD红外光谱可见,3 343 cm-1处为环糊精表面羟基的伸缩振动峰,2 921 cm-1处为环糊精CH2伸缩振动吸收峰。Fe3O4红外光谱中586 cm-1和3 300~3 500 cm-1处的宽峰分别为Fe―O的特征吸收峰及其表面羟基的伸缩振动峰。Fe3O4@SiO2和Fe3O4@SiO2@β-CD存在1 105 cm-1和1 111 cm-1处拉伸的特征硅峰Si―O―Si,表明二氧化硅外壳已形成。此外,Fe3O4@SiO2和Fe3O4@SiO2@β-CD的其他特征吸收带Si―OH拉伸振动吸收峰、Si―O弯曲振动吸收峰、Si―O―Si弯曲振动吸收峰分别为960、795、475 cm-1和958、794、474 cm-1,Fe―O的特征峰从586 cm-1移至582 cm-1,表明二氧化硅壳通过Fe―O―Si化学键连接到磁性Fe3O4纳米颗粒表面。Fe3O4@SiO2@β-CD的光谱图中,2 850 cm-1和2 924 cm-1处存在信号增强的不对称和对称的C―H伸缩振动吸收峰,且峰的重叠导致信号增强,表明β-CD与Fe3O4@SiO2已成功接枝形成Fe3O4@SiO2@β-CD材料。
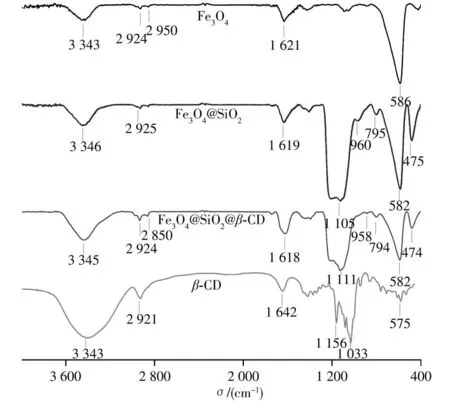
图3 β-CD、Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD的红外光谱图Fig.3 FT-IR spectra ofβ-CD,Fe3O4,Fe3O4@Si O2 and Fe3O4@SiO2@β-CD
2.1.3 X射线衍射表征β-CD、Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD吸附材料进一步采用X射线衍射(XRD)表征(图4)。结果显示:磁性Fe3O4在29.2°、35.6°、43.4°、53.5°、57.1°和62.7°出现特征衍射峰,分别对应Fe3O4的(220)、(311)、(400)、(422)、(511)、(440)晶面的衍射峰(JCPDS No 19-0629),当涂覆SiO2后,在16°~26°处出现非晶体SiO2导致的微弱宽衍射峰,无尖锐的衍射峰,这是因为涂覆过程采用的是TEOS,因而在Fe3O4表面生成的是无定形硅基材料,所以SiO2衍射峰不明显,仅在18.14°出现对应的非晶体态硅壳的弱衍射峰[28]。进一步在Fe3O4@SiO2表面键合环糊精,于6.1°处出现β-CD的有关特征峰,表明环糊精已成功键合在Fe3O4@SiO2表面。
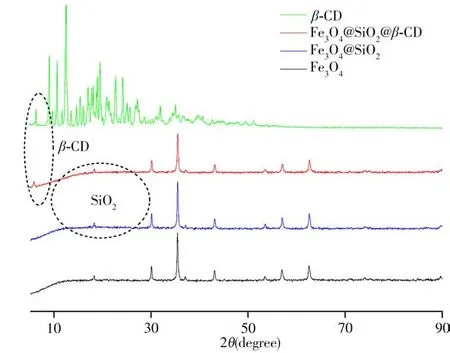
图4 β-CD、Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD的XRD光谱图Fig.4 XRD spectra ofβ-CD,Fe3O4,Fe3O4@SiO2 and Fe3O4@SiO2@β-CD
2.1.4 N2吸附-脱附表征 由Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD复合材料样品在77 K下的N2吸附等温线和孔径分布图可见(图5A),三者均含有明显的滞后环,这是典型的Ⅳ型等温线,当相对压力(P/Po)小于0.1时,材料对氮气的吸附量急剧增加,表明在较低压力下,复合材料和氮气的相互作用力大,Fe3O4@SiO2@β-CD等复合材料含有微孔,而回滞环的存在可能是由于Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD纳米材料尺寸太小(≈200 nm)且具有磁性作用,使得纳米粒子间相互吸附产生中孔和大孔的形状结构,从而与氮气发生相互作用所致。从Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD复合材料的孔径分布图和不同孔径的分布情况可见(图5B、C),在Fe3O4上逐步涂敷SiO2和β-CD,合成的Fe3O4@SiO2和Fe3O4@SiO2@β-CD孔隙尺寸变大,3种材料的大孔比例分别为49.42%、49.85%和98.17%,其比表面积分别为25.044、21.046、15.374 m2/g,孔体积分别为0.134、0.745、0.993 mL/g,平均孔径分别为1.143、2.141、2.582 nm,由此可见,Fe3O4逐步涂敷SiO2和β-CD后,平均孔径进一步增加。因此,更大的孔可能使Fe3O4@SiO2@β-CD相对于Fe3O4@SiO2和Fe3O4能更好地吸附目标分析物。
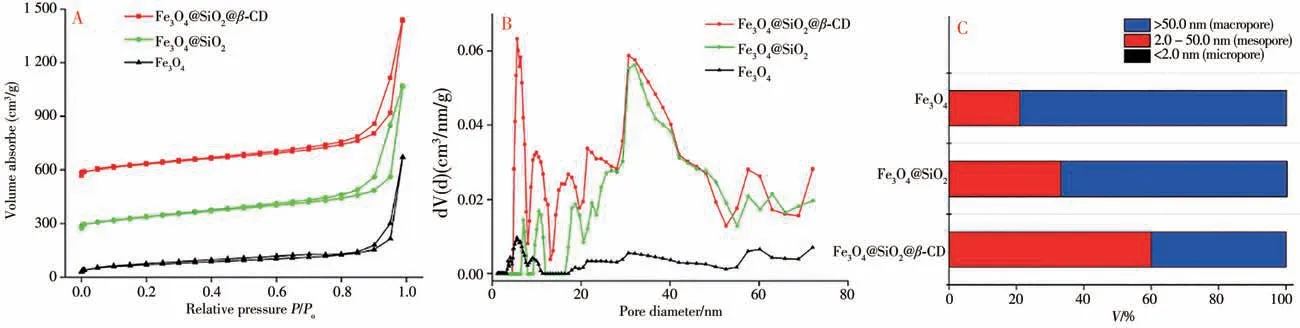
图5 Fe3O4、Fe3O4@Si O2和Fe3O4@Si O2@β-CD的氮气吸附-脱附等温线和孔径分布图Fig.5 Nitrogen adsorption-desorption isotherm and pore size distribution map of Fe3O4,Fe3O4@SiO2 and Fe3O4@SiO2@β-CD
2.2 萃取条件的优化
为进一步提高吸附剂的萃取效率,实验优化了萃取时间、解吸时间、解吸溶剂、萃取液pH值和盐离子浓度等影响因素。将50 mg Fe3O4@Si O2@β-CD加入50 mL含有100μg/L联苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯的混合水溶液中,通过回收率及解吸率参数考察材料的吸附萃取性能,具体公式为:E=m1/(m1+m2+m3+m4+…)×100%;R=me/m0×100%;式中E为解吸率,m1为第一次解吸后目标物的质量(μg),m2是第二次解吸后目标物的质量(μg),依次解吸直至无目标分析物检出;R为萃取率,me为吸附后目标物的质量(μg),m0为吸附前溶液中目标物的质量(μg)。
2.2.1 萃取吸附剂的选择 分别采用制备的Fe3O4@SiO2@α-CD、Fe3O4@SiO2@β-CD和Fe3O4@SiO2@γ-CD萃取100μg/L的联苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯混合水溶液,考察了3种环糊精衍生物(α-CD、β-CD和γ-CD)复合材料吸附性能的差异。结果显示:3种环糊精磁性材料均能吸附4种拟除虫菊酯类化合物,但键合β-CD的吸附剂对所有分析物的萃取率最大,因此选择Fe3O4@SiO2@β-CD为磁性固相萃取吸附剂。
2.2.2 萃取实验参数的优化 实验考察了萃取时间、解吸时间、解吸溶剂、萃取溶液pH值及离子强度对Fe3O4@SiO2@β-CD吸附4种拟除虫菊酯类物质的影响。结果显示,改性后的磁性材料可在30 min内对4种物质达到吸附平衡,因此选择最佳萃取时间为30 min。进一步研究了Fe3O4@SiO2@β-CD材料在不同时间(2~20 min)超声解吸对4种拟除虫菊酯类物质萃取效率的影响,发现均可在15 min达到解吸平衡。另外,由于拟除虫菊酯类物质在不同解吸溶剂中有不同的分配系数,因此实验考察了甲醇、乙腈、乙醇、异丙醇、二氯甲烷5种不同解吸溶剂对萃取效率的影响。结果显示:当解吸溶剂为甲醇时,联苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯有最大的萃取效率,因此选择甲醇为最优解吸溶剂。由于目标分析物在不同pH值溶液中存在的形态不同,从而影响分析物的萃取效率,因此实验考察了不同pH值(2.0、4.0、6.0、7.0、8.0、10.0、12.0)对萃取效率的影响,发现在酸性或碱性条件下,萃取效率明显下降,这可能是由于拟除虫菊酯类物质在过酸过碱的水溶液中解离,从而导致萃取效率下降。因此,萃取溶液的最佳pH值选为7.0。在提取过程中加入适量NaCl也会影响提取效率,且4种拟除虫菊酯类物质的萃取效率随离子强度的增加而下降,这是因为磁性复合材料中β-CD的空腔对Na+也有一定包结作用,使得Fe3O4@SiO2@β-CD与4种拟除虫菊酯类物质的相互作用力减弱,导致吸附效率降低。因此,萃取液中不添加NaCl更有利于材料对目标物的吸附分离。
2.2.3 萃取容量 实验通过萃取容量考察了Fe3O4、Fe3O4@SiO2和Fe3O4@SiO2@β-CD的吸附能力,在最佳萃取条件下,萃取质量浓度为100~4 000μg/L的拟除虫菊酯混合溶液,其萃取量为Q/(C0-CR)×V/M,式中Q为分析物的萃取量,C0为解吸后得到的质量浓度(μg/L),CR为吸附前溶液中目标物质量浓度(μg/L),V为解吸后的进样体积,M为Fe3O4@SiO2@β-CD复合材料的质量(g)。结果显示,未经修饰的裸露四氧化三铁由于表面相对光滑,与拟除虫菊酯类物质作用的活性位点相对较少,因此吸附量较低;而在Fe3O4表面涂敷二氧化硅材料后使得材料表面增加了可吸附拟除虫菊酯类物质作用的活性位点,因此Fe3O4@SiO2对4种拟除虫菊酯类物质的吸附量有所增加;继续键合β-CD后形成的Fe3O4@SiO2@β-CD的环糊精的空腔结构与拟除虫菊酯类化合物的分子大小接近,进一步增加了对拟除虫菊酯类物质的作用位点,因此Fe3O4@SiO2@β-CD复合材料对拟除虫菊酯类物质的吸附量远大于未经修饰的Fe3O4和Fe3O4@SiO2。
2.3 分析方法的建立
以Fe3O4@SiO2@β-CD为磁固相萃取吸附材料,在优化条件下采用GC-MS分析了联苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯4种拟除虫菊酯类杀虫剂。结果显示:在0.1~3.0 mg/kg含量范围内,4种拟除虫菊酯类杀虫剂的响应信号与对应浓度呈良好的线性关系,相关系数(r2)为0.998 1~0.999 8,以3倍信噪比(S/N≥3)计算得其检出限(LOD)均为0.03 mg/kg,以S/N≥10计算得定量下限(LOQ)均为0.1 mg/kg(表1),完全满足烟叶中4种拟除虫菊酯检测灵敏度要求,总离子流图见图6。

表1 4种拟除虫菊酯类农药的线性方程、相关系数(r2)、检出限和定量下限Table 1 Linear equations,correlation coefficients(r2),LODs and LOQs of four pyrethroid pesticides
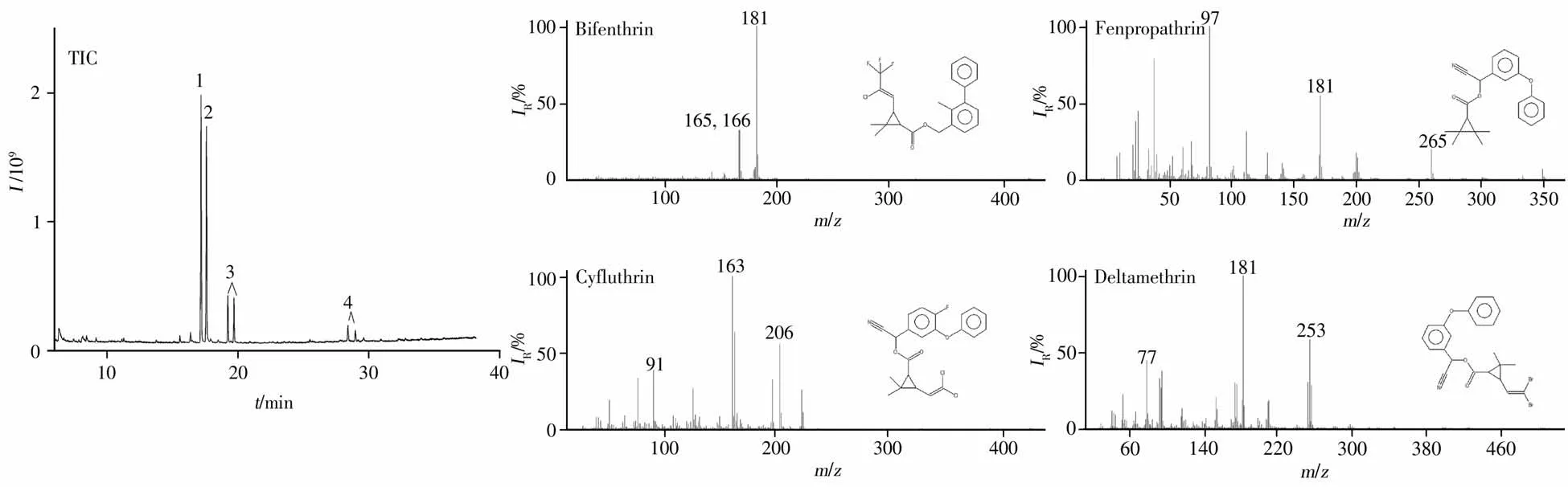
图6 4种拟除虫菊酯总离子流图及其质谱图Fig.6 Total ion chromatogram and mass spectra of four pyrethroids 1:bifenthrin,2:fenpropathrin,3:cyfluthrin,4:deltamethrin
2.4 实际样品分析及加标回收率
采用本方法在优化条件下对10批市售烟叶样品进行分析,结果在5批烟叶样品中检出联苯菊酯、氟氯氰菊酯和溴氰菊酯农药残留,含量分别为1.08~1.78、0.10~0.35、0.12~0.75 mg/kg,但均未检出甲氰菊酯。根据2020年国际烟草科学研究合作中心(CORESTA)农药限量名单和2006年农用化学品咨询委员会(ACAC)提出的现行法规中烟草中农药的最大残留限量标准(联苯菊酯、氟氯氰菊酯和溴氰菊酯限量分别为3.00、2.00、1.00 mg/kg)[29],检出的联苯菊酯、氟氯氰菊酯和溴氰菊酯均未超标,其中一个阳性烟叶样品色谱图见图7。
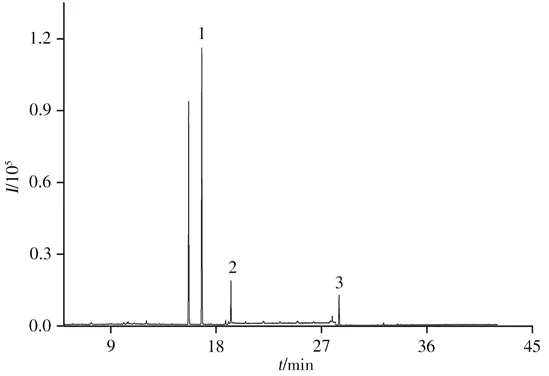
图7 阳性烟叶样品的选择离子流图Fig.7 Selected ion flow diagram of a positive tobacco sample 1:bifenthrin,2:cyfluthrin,3:deltamethrin
取另外5批阴性烟叶样品(烤烟、白肋烟、香料烟、晒红晒烟和晒黄晒烟)分别添加一定量4种拟除虫菊酯类杀虫剂,在优化条件下测定,计算方法的回收率及相对标准偏差(RSD)。结果显示:4种拟除虫菊酯类杀虫剂的回收率为65.9%~107%,RSD为0.70%~12%(表2)。表明方法具有较好的准确性和精密度,可用于实际烟叶样品中联苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯4种拟除虫菊酯类杀虫剂的检测。
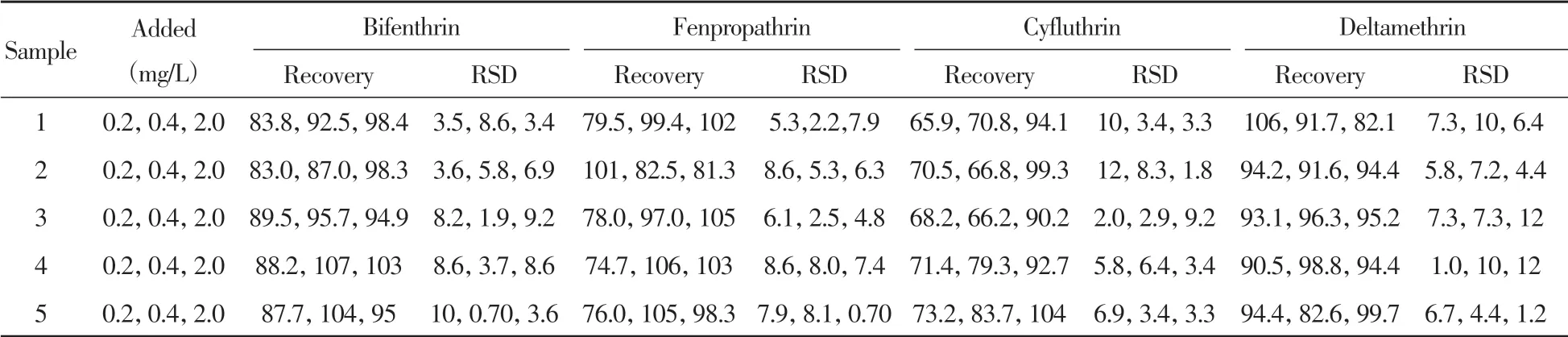
表2 不同烟草中4种拟除虫菊酯类杀虫剂的回收率的测定(n=3)Table 2 Determination of recovery of four pyrethroid pesticides in different tobaccos(n=3) (%)
3 结 论
本文将环糊精结合磁性纳米Fe3O4颗粒得到具有磁性的环糊精磁性复合材料,并联用气相色谱-质谱建立了烟叶中联苯菊酯、甲氰菊酯、氟氯氰菊酯和溴氰菊酯4种拟除虫菊酯类杀虫剂的快速分析方法,方法的检出限为0.03 mg/kg,定量下限为0.1 mg/kg,回收率为65.9%~107%,RSD为0.70%~12%。该环糊精磁性复合材料具有制备简单,样品前处理易于操作,萃取效率高且易分离等优点,可用于烟叶样品中拟除虫菊酯类杀虫剂农药残留的检测分析。
猜你喜欢
食品安全导刊·中旬刊(2022年3期)2022-04-15
纺织科技进展(2021年3期)2021-06-09
环境与发展(2019年6期)2019-08-06
分析化学(2019年3期)2019-03-30
农业工程技术·温室园艺(2017年10期)2017-12-02
家庭医学(2017年3期)2017-05-27
现代农业科技(2016年20期)2016-12-20
哈尔滨理工大学学报(2015年4期)2015-12-31
分析化学(2015年7期)2015-07-30
山东农药信息(2013年10期)2013-11-27
