
辉光放电质谱法定量测定重掺晶体硅中替位碳含量
2021-08-05 08:31李朋飞杨复光刘鹏宇胡芳菲赵景鑫
中国无机分析化学 2021年4期
刘 红 李朋飞 刘 英 杨复光 刘鹏宇 胡芳菲 赵景鑫
(1.国标(北京)检验认证有限公司,北京,100088;2.陕西有色天宏瑞科硅材料有限责任公司,陕西 榆林 719000;3.国合通用测试评价认证有限公司,北京 100088)
前言
硅中的碳是一种有害杂质,碳的存在会减小硅的晶格参数,降低少数载流子的寿命,使器件的击穿电压大大降低,漏电流增加。碳的不均匀分布会导致硅片的条纹,并会导致硼的不均匀分布[1-2],因此应尽量避免碳杂质的引入。但在晶体硅提纯过程中由于原料、设备等原因经常带入碳,为了有效控制碳含量提高器件成品率[3-4],碳杂质含量检测尤为重要。
目前硅中碳常用的检测手段有二次离子质谱法(SIMS)[5]、熔化分析法(GFA)[6]、红外吸收光谱法[7-9],何友琴等用SIMS测定了硅中碳和氧,SIMS属于半定量分析,需要标准样品校准仪器[10-11],而目前还没有市售的晶体硅标准样品,有些实验室采用离子注入法制备了硅参照样,离子注入设备复杂而且昂贵,一般实验室还不具备该项能力;另由于一次离子溅射源和设备参数对离子产率影响较大,因此获得高灵敏度需要较长时间的调试。红外吸收光谱法测量硅中替位碳方便精确、重复性好,对样品无破坏,但对于重掺后电阻率低于0.1 Ω·cm 的晶体硅样品,杂质的吸收峰淹没于自由载流子的吸收峰中,导致碳的测量无法实现。辉光放电质谱法[12-14]采用固体直接进样、具有灵敏度高、线性范围宽、检测速度快、不受杂质化学形态影响等特点而广泛应用于电子、冶金、航空航天等领域材料的检测。
低温傅里叶变换红外光谱法(LT-FTIR)具有测定速度快、结果重复性好、准确度高等优点,广泛用于单晶硅中碳的测定[7],并常作为溯源手段。本文利用低温傅里叶红外光谱法对4个参考样中替位碳进行多次测定,每个点多次测定数据稳定后平均值作为该硅片中替位碳的实际含量,用该系列参考样校准辉光放电质谱仪(GDMS)的相对灵敏度因子,从而达到定量测定重掺晶体硅中替位碳的目的。
1 实验部分
1.1 主要仪器
Nu Astrum辉光放电质谱仪(英国Nu Instruments仪器公司);CryoSAS低温傅里叶变换红外光谱仪(德国Bruker);切割磨抛两用台锯切割机(1 280 W,转速23 000 r/min)。
1.2 试剂
硝酸(UP级),氢氟酸(UP级),混酸蚀刻液(硝酸:氢氟酸=4∶1),稀氢氟酸(氢氟酸:水=1∶100),高纯氩气(≥99.9999%),压缩空气(0.6 MPa),高纯氮气(≥99.999%)。
1.3 样品前处理
用区熔法拉制单晶硅棒[15],在方法和装置固定情况下,熔区长度保持不变。通过测量掺杂曲线可以确定杂质沿晶棒长度方向的浓度分布。沿长度方向将晶棒切成硅片,测量每个硅片的碳含量,然后绘制浓度与熔区长度的分布曲线,见图1。以熔区长度为15 mm的区熔方式,碳的有效分凝系数为0.175,在12倍熔区长度处分别切硅片和硅棒,见图2;硅片(a1、b1、c1、d1)用作LT-FTIR测量碳含量,硅棒(a2、b2、c2、d2)用作GDMS校准仪器的RSF。
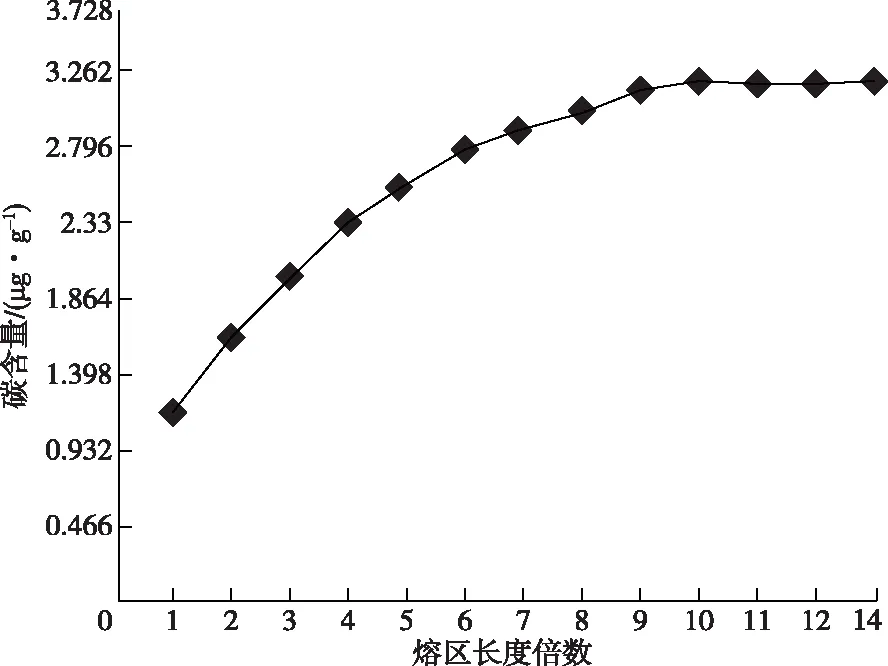
图1 碳的轴向含量分布曲线Figure 1 Axial distribution curve of carbon content.
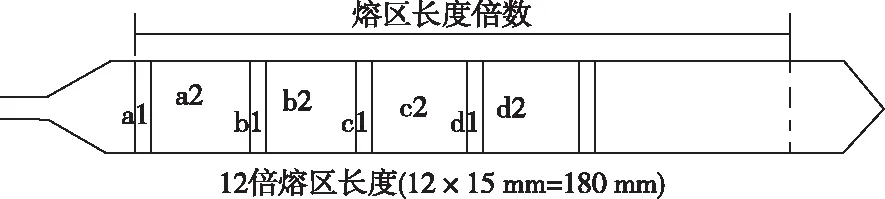
图2 熔区长度测量和晶棒取样位置Figure 2 Measurement of melting zone length and sampling position of crystal bar.
参照样a1、b1、c1、d1,将单晶硅片加工成直径为11 mm、厚为2 mm的双面抛光圆片用于低温红外吸收光谱法测定。
单晶硅棒母材均匀性满足要求前提下,对应低温傅里叶红外用参照样片,分别从4个硅棒取样a2、b2、c2、d2,加工成3 mm×3 mm×25 mm的棒状,用于辉光放电质谱法测定
待测样x:选择重掺Sb的样品x,Sb含量为120 μg/g,用作验证GDMS法准确度和精密度,制成3 mm×3 mm×25 mm的棒状。
样品清洗:用混酸腐蚀样品,至少腐蚀两次,使样品表面除去不少于100 μm的厚度,然后用超纯水冲洗至少3遍,用稀氢氟酸于120 ℃浸泡10 min,然后用去离子水超声清洗10 min,洗干净的硅棒用高压氮气吹扫待测。
1.4 样品测定
通过对放电电压、放电电流、放电气体流量进行优化,得到最佳工作条件,见表1。选择7个可覆盖低、中、高质量数具有代表性的元素对影响信号峰形状、分辨率的Quad Values进行调谐,此时碳信号峰形对称,强度符合检测灵敏度需求,分辨率满足干扰分离要求,见表2。

表1 辉光放电质谱仪工作条件

表2 Quad 参数值
2 结果与讨论
2.1 放电参数
2.1.1 放电电流
考察放电条件对Si放电的影响,随着放电电流的增加,Si的离子化率随之增加,信号强度逐渐加大。当电流增加至2.5 mA后,正负极间Si原子的电离、带电粒子的碰撞、迁移达到稳定状态,带电粒子密度不再有明显波动,在一定范围内电场不足以提供更多的能量满足Si+的二次电离,而长时间过大的电流会导致大量样品离子在放电舱内的沉积,从而降低仪器灵敏度,因此选择放电电流为2.5 mA。
2.1.2 放电电压
辉光放电作为离子源,是在电场作用下带正电离子轰击样品表面,使得样品原子获得能量后脱离样品表面进入等离子体,中性原子扩散进入负辉区离子化,当正负极间电压持续增加时,带电粒子的移动速度增加,在一定程度上会增加放电电流,样品原子获得能量后加快电离,信号强度呈增强趋势,当电压增加到一定阶段,带电粒子密度达到饱和状态,电流不再增加,信号强度不再有明显变化。设定放电电流为2.5 mA,放电电压从1 050 V增加至1 250 V时,Si信号强度从2.3×109cps增加至2.6×109cps,1 250 V以后曲线呈平台状,因此选择放电电压为1 250 V。
2.2 预溅射时间
酸腐蚀清洗很难彻底去除样品表面杂质,开始溅射后C信号强度缓慢下降,当溅射时间持续约35 min 后,C信号强度不再有明显波动,此时可进行采集数据,预溅射时间设置为35 min。
2.3 相对灵敏度因子
用LT-FTIR对单晶硅片a1、b1、c1、d1中替位碳含量分别进行6次测定,取6次结果的平均值作为该样片中替位碳的实际含量WLT,测定结果见表3。

表3 参照样中碳含量
在优化过的工作条件下,用GDMS逐个测定a2、b2、c2、d2中C与Si的离子束比值(IBRC/Si),每个样品测定6次,取其平均值作为该样品中碳的相对含量WGD(即IBRC/Si),将WGD作为横坐标,WLT作纵坐标,根据C=IBRC/Si×RSFC/Si,得到标准工作曲线y=ax+b,见图 3。a值即为RSFC/Si。统计4个点所得RSF精密度,相对标准偏差RSD为7.2%,精密度较好,见图4。
2.4 精密度实验
在优化过的工作条件下,重复测定Si中C,每两次之间将样品退出放电室,然后再重新装入,统计10次测定结果,离子束比值(IBR)与测定时间关系见图5,相对标准偏差RSD为2.9%,仪器稳定性较好,满足方法精密度要求。

图3 离子束比值与参照样中碳含量关系曲线Figure 3 Relation curve between ion beam ratio and carbon content in reference sample.
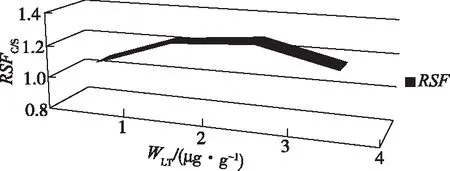
图4 RSFC/Si与参照样中碳含量关系曲线Figure 4 Relation curve between RSFC/Si and carbon content in reference sample.

图5 碳硅离子束比值与测定时间关系曲线Figure 5 Relation curve between the ratio of carbon-silicon ion beam and the measuring time.
2.5 准确度实验
根据GDMS结果计算原理,计算公式见式(1):
C=IBRC/Si×RSFC/Si
(1)
式中:C为待测杂质含量,%;
IBRC/Si为杂质与基体信号强度比(IBR=Ic/Isi);
RSFC/Si为碳的相对灵敏度因子。
在优化过的工作条件下将a2、b2、c2、d2作为未知样进行检测,利用RSFC/Si进行计算,得到碳含量记为Wcal,见表4。与低温红外光谱结果对照相对误差为不大于3.78%,两种方法测定结果一致性较好。
对待测样品进行测定,结果为3.16 μg/g,见表5。二次离子质谱法测得结果为3.04 μg/g,相对标准偏差为3.7%。

表4 辉光放电质谱法测定硅中替位碳结果

表5 样品检测结果
3 结论
辉光放电质谱法灵敏度高、检出限低、测定速度快、不受单晶硅片中重掺元素的影响,通过LT-FTIR定量测定参照样,校准GDMS的RSF,得到GDMS的定量结果,与红外光谱法和二次离子质谱法对照,相对误差<5%,结果一致性较好,是重掺单晶硅片中痕量替位碳测定的理想方法,对重掺晶体硅的质量控制具有重要意义。
猜你喜欢
现代经济信息(2022年26期)2022-11-16
阿来研究(2020年1期)2020-10-28
机电工程(2020年3期)2020-03-31
金刚石与磨料磨具工程(2019年4期)2019-09-18
能源(2018年10期)2018-01-16
能源(2016年2期)2016-12-01
光学精密工程(2016年2期)2016-11-07
太阳能(2016年6期)2016-09-23
大众文艺(2015年18期)2015-11-28
网印工业(2015年3期)2015-06-05
