
我国药品紧急使用的制度问题及对策建议*
2021-08-05 00:28徐东紫欧阳昭连
中国药业 2021年14期
陈 娟,卢 岩,徐东紫,张 婷,严 舒,欧阳昭连
(中国医学科学院医学信息研究所,北京 100020)
进入21世纪以来,全球遭遇了多次突发公共卫生事件,包括重症急性呼吸综合征(SARS)、中东呼吸综合征(MERS)、埃博拉病毒、寨卡病毒及新型冠状病毒肺炎(简称新冠肺炎)等疫情,这些事件给医疗系统和社会经济造成了巨大冲击,如何有效应对突发公共卫生事件是各国面临的重要议题[1]。一种新的突发公共卫生事件发生时,短时间内常缺乏经充分证实有效的可用药品,而研发一种新的药品需要较长时间,这就意味着必须选择一些已上市药品或在研药品紧急使用[2-3]。然而,紧急使用未上市药品和超药品说明书使用已上市药品在我国缺乏相应法律基础[4-5]。我国针对突发公共卫生事件建立的药品特别审批制度及当前已有的其他特殊审批通道均无法解决应急状态下的药品紧急使用问题[6-7]。美国针对突发公共卫生事件建立了药品紧急使用授权制度,且该制度经过十几年的不断完善已趋于成熟[8-9]。本研究中拟采用文献调研法和专家咨询法探讨我国新冠肺炎疫情期间使用未上市药品或超药品说明书用药的制度问题、我国现有特别审批制度及其他特殊审批通道在应对突发公共卫生事件中的不足,并借鉴美国药品紧急使用授权制度,为我国药品紧急使用授权制度的建立提供建议。现报道如下。
1 我国药品紧急使用的制度缺陷
1.1 新冠肺炎疫情涉及药物
治疗性药物:新冠肺炎疫情暴发以来,国家卫生健康委员会先后发布了多个版本的诊疗方案,其中推荐了多种治疗药物,包括未上市药品(如瑞德西韦),适应证或用法用量超药品说明书的已上市药品(如α-干扰素、洛匹那韦利托那韦、利巴韦林、氯喹、羟氯喹、阿比多尔、法匹拉韦、达芦那韦考比司他、奥司他韦、托珠单抗、乌司他丁、甘草酸制剂等),在患者救治中发挥了重要作用[10-12]。但其药品说明书中大多无新冠肺炎适应证,部分药品尚未上市[3,13]。
预防性疫苗:2020年6月29日,康希诺生物股份有限公司宣布,其与军事科学院军事医学研究院生物工程研究所联合开发的腺病毒载体疫苗(Ad5-nCoV)已于2020年6月25日获得中央军委后勤保障部卫生局颁发的军队特需药品批件,获准仅限于军队内部使用,当时该疫苗尚处于临床试验阶段,并未正式获批上市。2020年8月22日,国家卫生健康委科技发展中心主任、国务院联防联控机制科研攻关组疫苗研发专班工作组组长郑忠伟在央视《对话》栏目上介绍,我国已于2020年7月22日正式启动国药集团2款灭活疫苗的紧急使用,截至8月底已在医务人员、防疫人员、边检人员等特殊人群中接种了数十万人次,当时该疫苗同样处于临床试验阶段,并未正式获批上市。
1.2 相关法律法规
突发公共卫生事件应急状态下的药品使用应遵循的上位法,包括突发公共卫生事件应急相关法律法规和药品注册审评相关法律法规[14-21]。现对两类法律法规文件中与“药品应急”有关的条款进行如下梳理(见图1)。

图1 我国在新冠肺炎疫情期间使用未上市药品或超药品说明书用药应遵循的法律依据Fig.1 Legal terms that should be followed when using investigational drugs or off-label drugs during the COVID-19 epidemics in China
突发公共卫生事件应急相关法律法规:我国应对突发公共卫生事件的法律依据主要包括《中华人民共和国传染病防治法》(简称《传染病防治法》)、《中华人民共和国突发事件应对法》(简称《突发事件应对法》)和《突发公共卫生事件应急条例》[16-18]。《传染病防治法》于1989年2月21日通过,之后分别于2004年和2013年进行了修订,是我国应对传染病的主要法律依据。最新版中与药品应急有关的条款包括第四十九条和第六十三条,前者对传染病暴发时的药品生产和供应作了相应描述,后者对防治传染病的药品储备问题作了责任说明。《突发事件应对法》于2007年8月30日通过,自2007年11月1日起开始实施,该法律的颁布标志着我国突发事件应对正式进入法制化轨道。其中第三十二条对应急所需物资的监管、生产、储备、调拨和配送的权责分配作了相应说明。《突发公共卫生事件应急条例》于2003年5月9日颁布,成为我国应对突发公共卫生事件的主要依据,该条例于2011年进行了修订,最新版中与药品应急有关的条款包括第十六条、第十七条和第三十二条,上述条款对应急所需药品的储备、生产和供应问题作了说明(见表1)。然而,上述文件均未明确提及应急状态下的药品紧急使用问题。
药品注册审评相关法律法规:我国药品注册审评相关法律依据主要包括《中华人民共和国药品管理法》(简称《药品管理法》)、《中华人民共和国疫苗管理法》(简称《疫苗管理法》)、《药品管理法实施条例》和《药品注册管理办法》[14,15,20]。我国《药品管理法》于1984年通过,于2001年和2019年进行了2次修订,最新版《药品管理法》从2019年12月1日起开始实施,其中第二十三条、第二十六条和第九十六条分别对拓展性临床试验制度、附条件批准程序和优先审评审批程序进行了阐述,第九十二条对药品储备和紧急调用作了相关说明。我国《疫苗管理法》于2019年12月1日开始实施,其中第二十条指出,重大突发公共卫生事件急需的疫苗可以附条件批准,特别重大突发公共卫生事件所需疫苗可以在一定时间和范围内紧急使用;第二十八条规定,应对突发事件急需的疫苗可免于批签发。我国现行《药品管理法实施条例》是2002年根据旧版《药品管理法》颁布的,其中未提及药品紧急使用相关内容,新版《药品管理法实施条例》尚在制订中。我国《药品注册管理办法》于2002年通过,分别于2005年、2007年和2020年进行了修订,最新版《药品注册管理办法》于2020年7月1日起开始实施,其中第五十九条、六十三条、六十八条和七十二条分别对突破性治疗药物、附条件批准、优先审评审批和特别审批作了相关说明(见表1)。然而,上述文件仅《疫苗管理法》提及疫苗紧急使用,而治疗性药品的紧急使用尚缺乏明确的法律依据。

表1 当前法律法规文件中涉及药品应急或特殊审评通道的条款Tab.1 Provisions in current laws and regulations concerning drug emergency or special approval channels
2 我国现有审批制度应对突发公共卫生事件的不足
2.1 特别审批制度
2005年11月18日,原国家食品药品监督管理局审议通过了《国家食品药品监督管理局药品特别审批程序》[22]。所谓药品特别审批程序,是指在发生突发公共卫生事件时,对突发公共卫生事件应急处理所需药品进行特别审批。该程序是突发公共卫生状态下我国药品应急审批的主要依据,非突发公共卫生事件不能走该审批通道。即使是应对突发公共卫生事件的药品,也要经过严格筛选才能进入特别审批程序。如此次新冠肺炎疫情发生后,2020年1月22日至29日,国家药品监督管理局药品审评中心从企业提交的15件申请中筛选出7件进行专家组评估审核,仅通过审核3件,并报送国家药品监督管理局启动特别审批程序。
我国药品特别审批制度(审批程序见图2)不能成为突发公共卫生应急状态下使用未上市药品或超药品说明书用药的政策依据。该制度实际上是一种产品上市许可,仅加速了审批流程,但上市程序不减少,上市标准不降低,产品上市后需遵循上市后全生命周期管理的原则和规定,整个审批过程需耗费一定时间。但在突发公共卫生应急状态下,并无足够的时间等待产品通过特别审批后上市,因此新冠肺炎疫情期间,我国出现了使用未上市药品或超药品说明书用药的情况。
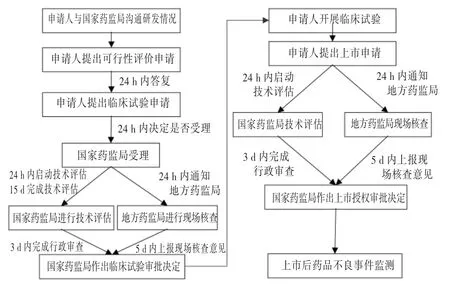
图2 我国药品特别审批程序Fig.2 Flow chart of special approval process of drugs in China
2.2 其他特殊审评通道或特殊用药制度
拓展性临床试验制度:是指将处于临床试验中、用于治疗严重危及生命且尚无其他治疗手段的疾病的药品用于未参与临床试验但病情相同的患者。该制度将使用人群限制在开展临床试验的医疗机构,且需由医师耗费一定时间申请才能获得,无法满足大批人群的需求[15]。
突破性治疗药物认定:是指在临床试验Ⅰ期或Ⅱ期阶段将具有明显临床优势的创新药或改良型新药纳入突破性治疗药物程序。该程序目的是加强沟通交流、加快临床试验进度,但在应急状态下,并不能达到尽快使用的目的[21]。
附条件批准程序:允许公共卫生方面急需的药品和应对重大突发公共卫生事件急需的疫苗在完成大部分研究程序后提前上市,属上市途径优化,但并不能达到足够快速使用的目的[21]。
优先审评审批程序:适用于防治重大传染病的创新药和改良型新药,也适用于疾病预防、控制所急需的疫苗。优先审评审批需在提出药品上市许可申请时提出,其目的是加快药品审评过程,鼓励创新药研发。即使进入优先审评通道,产品仍需历经上市所需的所有程序,并不能达到足够快速使用的目的[21]。
3 美国药品紧急使用授权制度经验总结
美国启动紧急使用授权(EUA)程序应对突发公共卫生事件[23-25]。EUA是指由美国食品和药物管理局(FDA)在紧急状态下对未获批准的医药产品及已获批准医药产品的未获批准用途进行授权。该制度经过10多年的发展,目前已较成熟,有明确的上位法、行业指南,相关文件中明确了启动条件、授权流程、产品范围、授权原则、授权期限和终止条件。
上位法:主要依据是《联邦食品、药品和化妆品法案》(简称《FD&C法案》)第564条,该条内容历经多次修订。2004年生效的《生物恐怖防疫计划法案》对《FD&C法案》第564条进行了修订,首次将医药产品紧急使用授权纳入法律范畴。2013年生效的《大流行与全风险防范再授权法案》(简称《PAHPRA法案》)对《FD&C法案》第564条作了进一步阐述,明确了紧急使用授权的产品范围包括药品、生物制品和医疗器械。2016年实施的《21世纪治愈法案》再次对《FD&C法案》第564条进行了修订,将产品范围进一步扩大,纳入动物用医药产品[24-25]。
行业指南:2007年7月,FDA公布了《医药产品紧急使用授权》文件,首次正式确定了EUA的制度程序。2017年1月,FDA发布《行业与其它利益攸关方行业指南:医药产品紧急使用授权与相关权限》,替代2017年发布的《医药产品紧急使用授权》文件[24-25]。
启动条件和授权流程:FDA启动紧急使用授权令的前提是国防部部长、国土安全部部长或卫生与公众服务部部长判断国内处于紧急状态。整个EUA程序涉及5个步骤,1)确定是否进入紧急状态(国防部、国土安全部或卫生与公众服务部);2)宣布进入紧急状态(卫生与公众服务部);3)审评EUA的请求(FDA);4)发布EUA或拒绝EUA的请求(卫生与公众服务部,但常会委托FDA代为发布);5)终止EUA(卫生与公众服务部)[24-25]。详见图3。
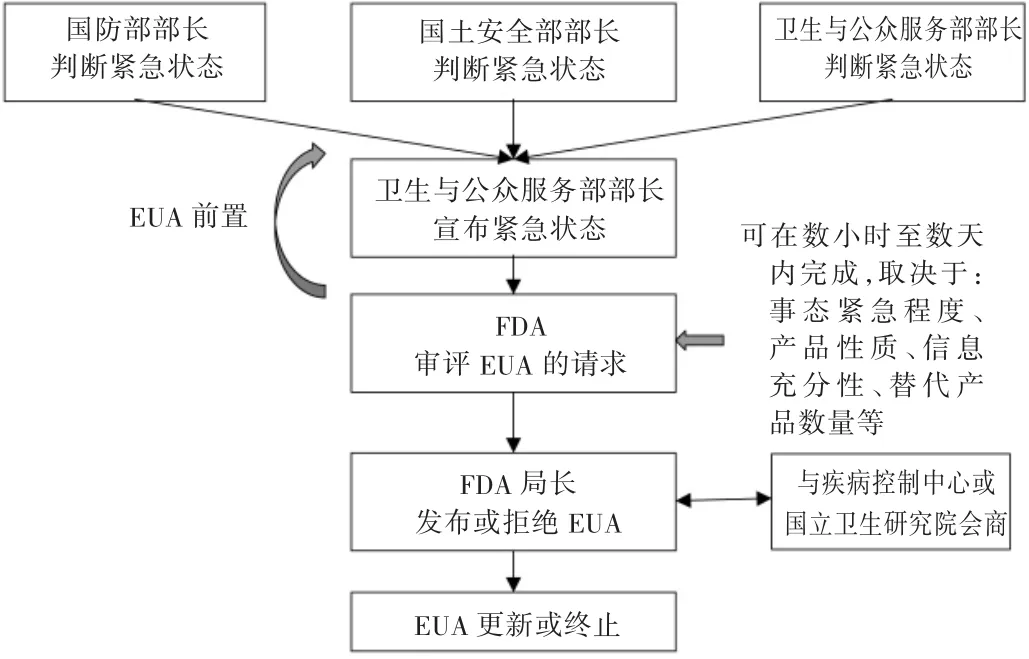
图3 美国药品紧急使用授权流程Fig.3 Authorization process of emergency use of drugs in the United States
产品范围及授权原则:潜在的EUA产品包括未获批准的医药产品及已获批医药产品未经批准的用途,此处的医药产品包括药品、生物制品和医疗器械。EUA不要求医药产品处于特定的研发阶段,但产品应正处于研发中,并完成了部分研发路径。EUA授权必须符合以下4个法定标准:1)事态紧急;2)产品有效;3)已知潜在获益大于潜在风险;4)无足够的已获批的替代产品。EUA可豁免部分监管要求,如知情同意或伦理委员会审批要求[23-25]。
授权期限和终止条件:EUA的授权期限为1年,当出现下列任一情况时,EUA随即终止。1)紧急状态终止时,EUA自动终止;2)EUA达1年期限时,卫生与公众服务部部长或受其委托的FDA局长决定是否延展或终止;3)经EUA授权的产品被FDA正式许可后,对产品的EUA即可作废[23-25]。
在H1N1流感和新冠肺炎疫情期间的应用经验:H1N1流感期间,FDA首次将EUA授予一个未上市的研究用药帕拉米韦,还将EUA授予磷酸奥司他韦和扎那米韦以扩大适应证。此后,美国EUA制度逐渐成熟,新冠肺炎疫情期间,FDA对多个药品授予了EUA,截至2020年底,瑞德西韦、新冠病毒中和抗体、2种组合疗法及2款新冠疫苗均已获得EUA,这些产品在应对新冠肺炎的流行中发挥了重要作用[23]。详见表2。
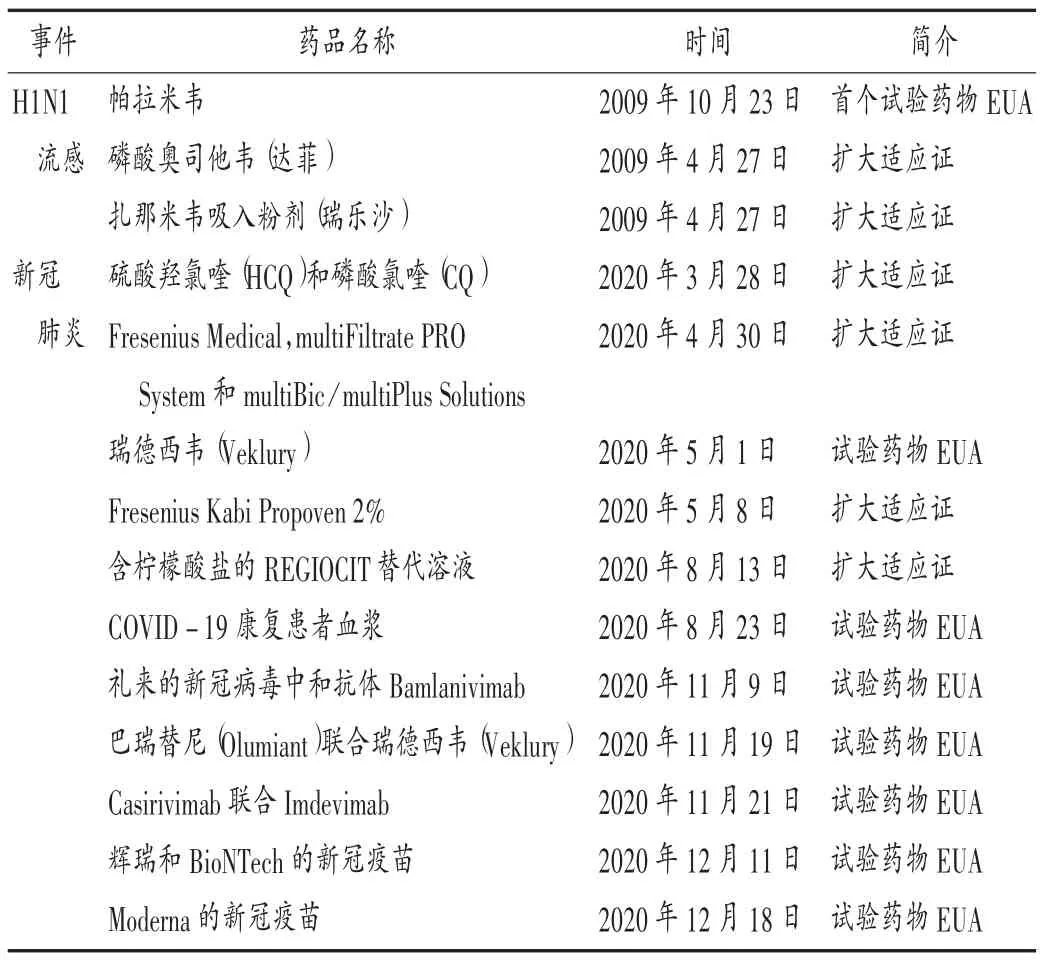
表2 美国应对H1N1流感和新冠肺炎期间授予的药品EUATab.2 Emergency-Use-Administration(EUA)of drugs granted by the United States in response to H1N1 influenza and COVID-19
4 建立我国药品紧急使用授权制度的建议
基于我国新冠肺炎疫情下药品紧急使用存在的问题,借鉴美国经验,提出如下建议。首先,完善药品紧急使用的上位法,目前紧急使用授权的产品范围仅包括疫苗,法律依据来自《疫苗管理法》,建议扩大紧急使用授权药物类型,在《药品管理法》中纳入治疗性药品紧急使用授权条款。其次,在《传染病防治法》《突发事件应对法》《药品管理法》《疫苗管理法》中明确紧急状态下如何启动紧急使用授权程序。再次,基于我国现有的药品特别审批制度,设计出药品紧急使用授权制度程序,建议该程序可适当简化,放宽标准,加速授权进程。最后,发布类似于行业指南的紧急使用授权指导性文件,在文件中明确紧急使用授权的启动条件、产品范围、授权原则、评估程序、授权期限、终止条件、责任豁免机制等内容。
猜你喜欢
中国合理用药探索(2022年1期)2022-11-26
广西医科大学学报(2022年5期)2022-06-07
煤气与热力(2021年3期)2021-06-09
父母必读(2021年3期)2021-02-04
金桥(2019年6期)2019-09-18
金桥(2018年10期)2018-10-09
金桥(2018年9期)2018-09-25
金桥(2018年7期)2018-09-25
小学生优秀作文(低年级)(2018年6期)2018-05-19
消费导刊(2017年20期)2018-01-03
