
胞外聚合物在腐败希瓦氏菌处理铀污染地下水中的作用
2021-08-02 03:31:42黄凤羽张海玲王永鹏易发成
原子能科学技术 2021年8期
黄凤羽,张海玲,王永鹏,王 哲,*,易发成,曾 铮
(1.西南科技大学 环境与资源学院,四川 绵阳 621010;2.中国工程物理研究院 材料研究所,四川 江油 621700)
铀矿开采和矿山尾矿会对邻近地下水造成铀污染,其铀含量高达50 mg/L[1]。由于铀的重金属毒性和放射性危害,含铀地下水的处理一直以来都受到广泛关注。传统的处理方式主要是物理化学方法(混凝沉淀法[2]、离子交换法[3]、吸附法[4]、膜分离法[5]等),在去除含铀地下水的同时通常有难以再生或二次污染等缺点[6-7]。微生物原位处理技术有原料充足、成本低、处理效率高、环境友好等优点,越来越受到人们的重视[8-10]。微生物对铀的去除已有广泛的研究,厌氧颗粒污泥[11]、硫酸盐还原菌、希瓦氏菌和假单胞菌等[12-14]都被证明对降低地下水中的铀浓度具有长期有效性。微生物固定铀的4个基本机制为:1) 微生物将U(Ⅵ)还原沉淀为U(Ⅳ);2) 细胞对U(Ⅵ)的吸收和积累;3) 微生物细胞表面对铀的吸附;4) 细胞释放的无机磷酸盐与U(Ⅵ)配位沉淀[15-17]。

1 实验
1.1 主要试剂与仪器
腐败希瓦氏菌(22940),中国工业微生物菌种保藏管理中心;DOWEX MARATHON C阳离子交换树脂(CER,20~50目)、乳酸钠(99.0%),美国Sigma-Aldrich;UO2SO4·3H2O(99.99%),湖北楚盛威化工有限公司,配制成2 200 mg/L U(Ⅵ)储备液。其他试剂均为市售分析纯,所有溶液均采用Milli-Q水制备。
模拟地下水溶液的配制参照文献[11],包括矿物元素和微量元素,具体如下:NH4HCO3,5 mg/L;K2HPO4,2 mg/L;MgCl2,2.1 mg/L;Ca(OH)2,1 mg/L;酵母提取物,0.33 mg/L;H3BO3,0.5 μg/L;FeSO4·7H2O,28 μg/L;ZnSO4·7H2O,1.1 μg/L;CuSO4·5H2O,1.6 μg/L;MnSO4·H2O,2.5 μg/L;(NH4)6Mo7O24·4H2O,2.0 μg/L;KAl(SO4)2·12H2O,1.75 μg/L;CoSO4·7H2O,23.6 μg/L;NiSO4·6H2O,1.13 μg/L;Na2SeO3·5H2O,1 μg/L;Na2WO4·2H2O,5.2 μg/L;EDTA,10 μg/L。将模拟地下水溶液压力灭菌20 min,冷却至室温后加入1 g/L NaHCO3溶液,将pH值调至7.0。U(Ⅵ)在地下水中的络合形态主要是碳酸氢盐结合态,因此用NaHCO3溶液作为模拟地下水溶液的缓冲溶液[13],最后充氮气10 min除去溶解氧。
NEXION 350电感耦合等离子体质谱仪、Nicolet-5700傅里叶变换红外光谱仪(FT-IR),美国Perkin Elmer公司;D3024台式高速微量离心机,美国赛洛捷克。
1.2 腐败希瓦氏菌培养
在无菌条件下将保存的腐败希瓦氏菌液按2%(体积分数)的接种量接到盛有培养基(组成为:胰蛋白胨,15 g;酵母提取物,5 g;氯化钠,5 g;水,1 L)的锥形瓶内,再置于恒温振荡器中(30 ℃,150 r/min)培养18 h,培养好的菌液以8 000 r/min离心5 min,之后用0.1 mol/L NaCl溶液洗涤2次。
1.3 腐败希瓦氏菌对U(Ⅵ)的固定
所有实验均在氮气条件下进行。具体步骤如下:1) 向50 mL血清瓶中加入20 mL模拟地下水溶液,再加入0.27 mL U(Ⅵ)储备溶液,使体系U(Ⅵ)最终浓度为30 mg/L;2) 在上述溶液中依次加入腐败希瓦氏菌和乳酸钠,浓度分别为6×108mL-1和10 mmol/L;3) 充高纯氮驱氧,用丁基橡胶塞和铝密封件密封,温度控制在(30±1) ℃;4) 在厌氧条件下定期用注射器从血清瓶中抽取0.5 mL菌体样液进行分析。反应结束后,取10 mL混合液进行EPS提取分析。每组实验均进行平行实验,并设置不加腐败希瓦氏菌的空白对照组。
1.4 分析方法
反应后腐败希瓦氏菌体中U(Ⅵ)和U(Ⅳ)的含量根据文献[24-25]的方法,采用NaHCO3-HNO3连续萃取法对腐败希瓦氏菌中固定的铀进行提取,其原理如图1所示。在该方法中,NaHCO3萃取的是吸附/络合的U(Ⅵ),而HNO3萃取的是不溶性U(Ⅳ)沉淀。在厌氧条件下定期用注射器从血清瓶中抽取0.5 mL菌体溶液,8 000 r/min离心分离5 min后,上清液过0.22 μm滤膜,记为U上清。剩余菌体细胞用厌氧0.1 mol/L NaCl溶液洗涤2次,去除菌体表面残留的U(Ⅵ)后,再加入0.5 mL 1 mol/L厌氧NaHCO3溶液提取菌体中的U(Ⅵ),厌氧条件下放置过夜后离心分离,上清液记为UNaHCO3。最后,再加入0.5 mL 10% HNO3提取菌体中的U(Ⅳ),放置4 h后离心分离,上清液记为UHNO3。所有样品均在5%HNO3中酸化保存,然后用ICP-MS测各组分浓度。
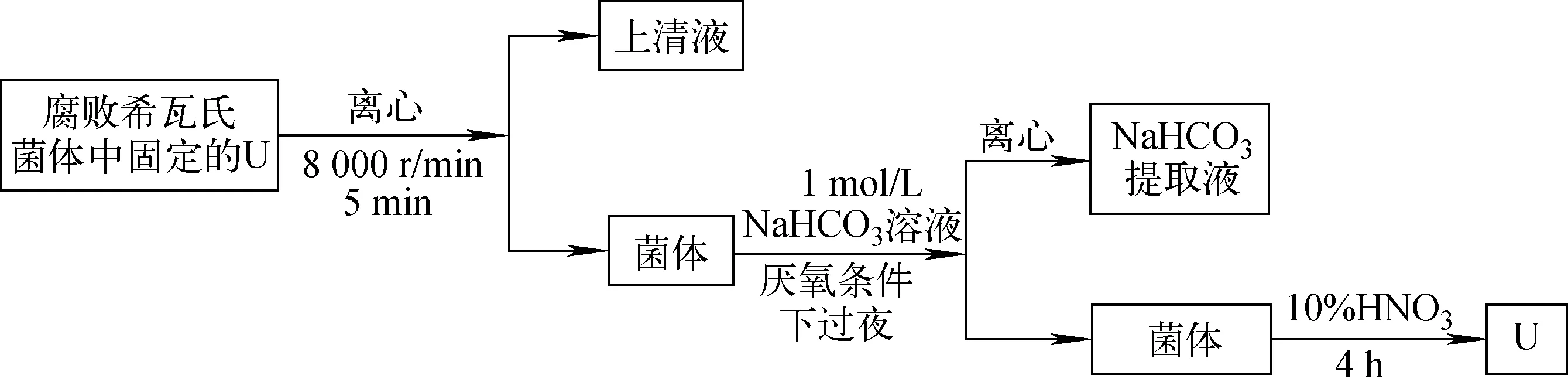
图1 腐败希瓦氏菌中U的NaHCO3-HNO3萃取法提取原理Fig.1 Extraction principle of U from Shewanella putrefaciens by NaHCO3-HNO3 fractionation method
1.5 EPS提取
采用阳离子交换树脂(CER)法、超声波法和离心法3种方法提取EPS。反应后的腐败希瓦氏菌用0.1 mol/L厌氧NaCl溶液(使用前煮沸并通入氮气驱氧)清洗2次,去除细胞表面的可溶性残留物质,再用0.1 mol/L厌氧NaCl溶液悬浮至10 mL。所有EPS提取过程均在厌氧条件下进行。
CER法:离子交换树脂可除去EPS和细胞之间的二价桥接离子,促使EPS从细胞表面脱附。取4 mL洗涤后的细胞悬浮液,按照70 g/g 的可挥发性悬浮物(VSS)标准称取CER量,600 r/min搅拌提取1 h,以14 510 r/min离心20 min,上清液过0.22 μm滤膜。
超声波法:超声波法提取EPS的原理是利用超声波产生的压力冲击使EPS和细胞分离。取4 mL洗涤后的细胞悬浮液,置于超声清洗器(150 W)中提取10 min,14 510 r/min离心20 min,上清液过0.22 μm滤膜。
离心法:离心法提取EPS的原理主要是利用高速离心产生的重力场作用,使EPS和细胞分离。取2 mL洗涤后的细胞悬浮液,以14 510 r/min的速度离心20 min,上清液过0.22 μm滤膜。
1.6 SP-ICP-MS分析EPS中的U
用单颗粒ICP-MS(SP-ICP-MS)对过0.22 μm滤膜后的EPS提取液中的可溶性U和颗粒U进行定量分析,并对颗粒U的粒径分布进行分析。在单颗粒模式下,可溶性铀酰离子的信号与含铀粒子的信号明显不同。因此,EPS中矿物U的含量为总U浓度与可溶性U浓度的差值。为表征纳米级铀粒子的粒径分布,用已知粒径和颗粒浓度的纳米银(nanoComposix公司)作为标准粒子。单个胶体粒子进入后会在等离子体焰矩内被粒子化为离子簇,以瞬时信号的形式被质谱检测到。其中信号强度反映颗粒粒径,信号频率反映颗粒浓度。
1.7 EPS反应前后FT-IR分析
将反应前后的EPS冷冻干燥后进行FT-IR分析,采用KBr压片法,扫描波数为400~4 000 cm-1,扫描次数为10,分辨率为4 cm-1。
2 结果与分析
2.1 腐败希瓦氏菌对U(Ⅵ)的固定
在腐败希瓦氏菌固定U(Ⅵ)反应过程中,对上清液中U的浓度(U上清)进行测定,并分析实验各阶段菌体中的U含量(U菌体=UNaHCO3+UHNO3)和系统的物质平衡(U系统=U上清+U菌体),结果示于图2。UNaHCO3和UHNO3分别为吸附的U(Ⅵ)(如U(Ⅵ)离子、U(Ⅵ)-碳酸盐矿物)和还原的U(Ⅳ)的浓度。由图2可知,未加腐败希瓦氏菌的空白组的U含量没有明显变化,说明U在实验体系中没有发生沉淀。上清液中U(Ⅵ)浓度随时间的增加不断下降,在0~28 h内腐败希瓦氏菌对铀的去除速度最快,之后逐渐趋于平稳,最终的去除率为90.9%。由图2还可看出,系统的U含量(U系统)在反应过程中没有明显变化,表明溶液中的U被腐败希瓦氏菌固定,系统有很好的物质平衡。由于碳酸氢盐是地下水系统中广泛存在的无机配体[26],所以本文着重模拟地下水碳酸氢根的体系。体系的初始pH值设定为7.0,但实验中发现,pH值随反应的进行因腐败希瓦氏菌消耗底物而有所下降,从7.0下降至6.2,处于弱酸性。对于酸度更大的环境(pH<6),这些材料可能也会适用,后续会进行相关研究。
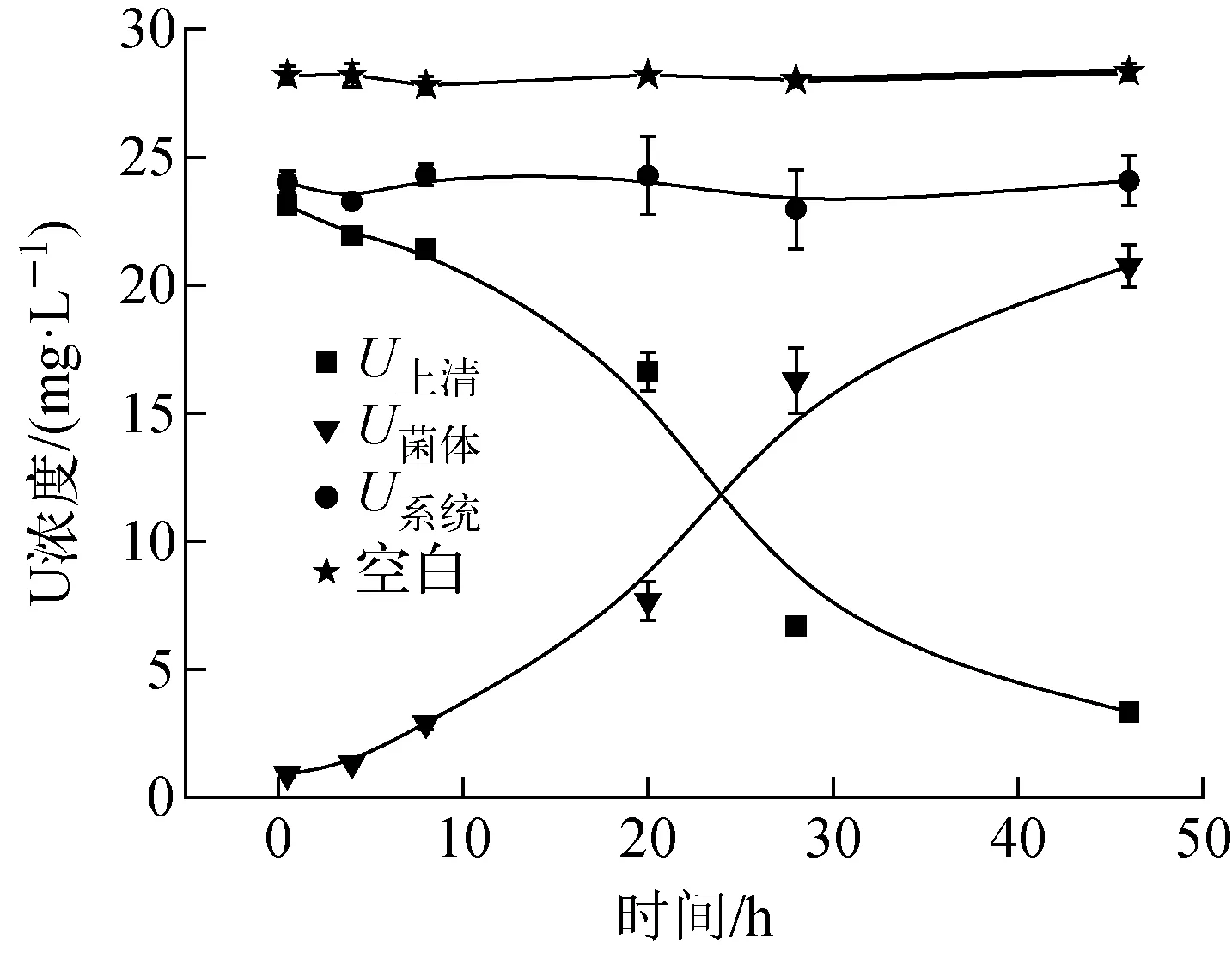
图2 腐败希瓦氏菌固定U(Ⅵ)过程中U上清、U菌体和U系统随时间的变化Fig.2 Usupernatant, Ucells and Usystem in U(Ⅵ) immobilization process by Shewanella putrefaciens
NaHCO3-HNO3连续提取反应过程中腐败希瓦氏菌体的U(Ⅳ)和U(Ⅵ)含量的变化示于图3。由图3可知,UNaHCO3和UHNO3随时间的增加而增加,说明腐败希瓦氏菌至少可通过吸附/络合和还原两种方式固定U(Ⅵ)。菌体中的铀含量由最初的(0.89±0.09) mg/L增加到(20.70±0.71) mg/L,与上清液中U减少的浓度大体一致,表明溶液中的U(Ⅵ)已被腐败希瓦氏菌固定。在初始厌氧条件下(t<8 h时),UHNO3较低,这是由于腐败希瓦氏菌在反应初期对缺氧体系的适应性调整,还原率较低,而随着反应的进行表现出较好还原效果。UHNO3从最初的(0.11±0.03) mg/L增加到反应结束时的(4.02±0.38) mg/L。以上结果表明,腐败希瓦氏菌除吸附作用外还具有还原U(Ⅵ)的能力,可有效将地下水中的U(Ⅵ)还原为U(Ⅳ)。
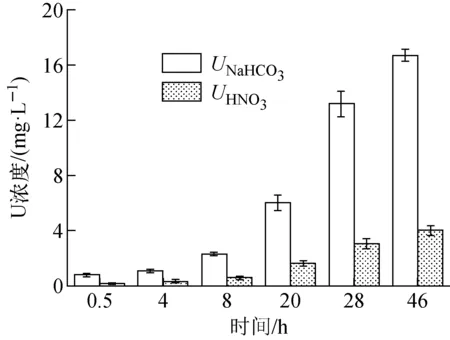
图3 腐败希瓦氏菌固定U(Ⅵ)过程中UNaHCO3和UHNO3随时间的变化Fig.3 UNaHCO3 and UHNO3 in U(Ⅵ) immobilization process by Shewanella putrefaciens
反应过程菌体中U(Ⅳ)/U(Ⅵ)比的变化示于图4。U(Ⅳ)/U(Ⅵ)比随时间的推移逐渐增加,表明随着反应的进行,腐败希瓦氏菌对U的还原活性逐步被激发。此外,还说明大多数U(Ⅵ)在还原成U(Ⅳ)之前先与腐败希瓦氏菌发生吸附反应,U(Ⅵ)与腐败希瓦氏菌的吸附反应较生物还原反应快得多。
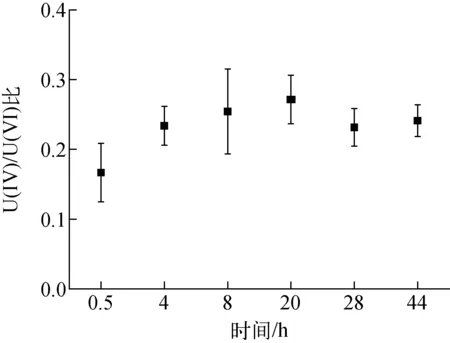
图4 腐败希瓦氏菌固定U(Ⅵ)过程中U(Ⅳ)/U(Ⅵ)比的变化Fig.4 Variation of ratio of U(Ⅳ)/U(Ⅵ) in U(Ⅵ) immobilization process by Shewanella putrefaciens
2.2 EPS中的铀含量
从腐败希瓦氏菌中提取的EPS中U的含量在很大程度上取决于提取过程中EPS的提取效率和不溶性U(Ⅳ)的再溶解程度。采用CER法、超声波法和离心法3种方法提取的EPS中U的含量和UEPS/TUcells比示于图5。从图5可看出,离心法和超声法所得EPS中铀含量(UEPS)较低,离心法中UEPS为(0.42±0.09) mg/L,超声法中UEPS为(0.85±0.15) mg/L,这一结果与EPS提取效率[27]一致,由于其对EPS提取效率较低导致UEPS的含量低。对于CER法,UEPS在很大程度上取决于萃取时间。但随着萃取时间的延长,不溶性U(Ⅵ)的再溶解也变得严重,在本研究小组之前的研究中,当萃取时间为1 h、转速为600 r/min时,U(Ⅵ)再溶解的程度是可接受的[24],CER法中UEPS为(2.91±0.22) mg/L,占腐败希瓦氏菌固定总铀量(TUcells=(20.7±0.71) mg/L)的(14.0±1.0)%。对比3种方法,离心法和超声法的铀提取效率低,不适用于EPS中铀的种类和含量的测定,CER法得到的结果可能更接近EPS中的实际铀含量。以上结果表明,用腐败希瓦氏菌固定铀时,EPS具有很强的富集能力,EPS中的铀含量不可忽略。
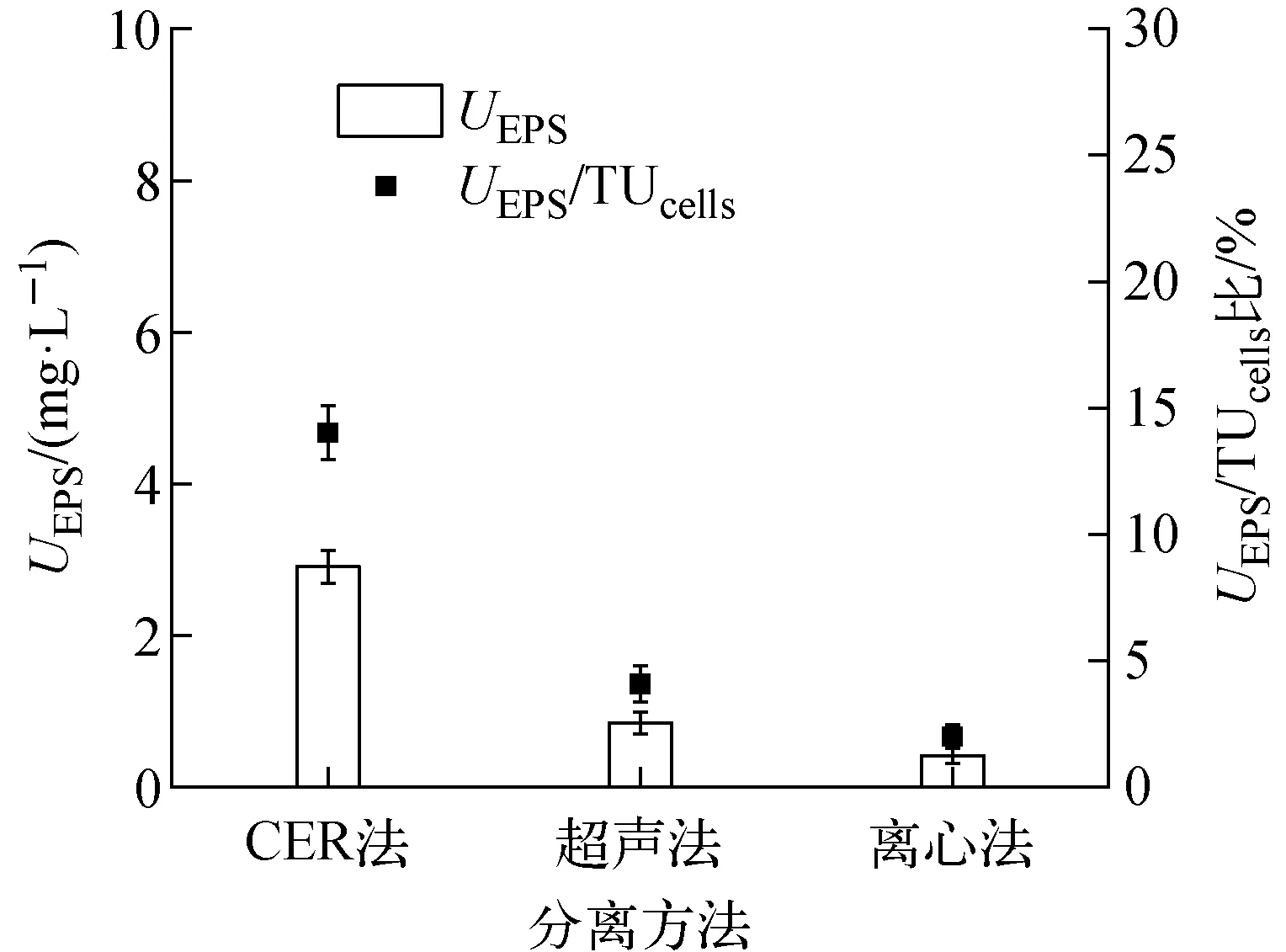
图5 从腐败希瓦氏菌中提取的EPS中的铀含量和UEPS/TUcells比Fig.5 UEPS and UEPS/TUcells ratio of EPS solution extracted from Shewanella putrefaciens
2.3 EPS中的颗粒铀
CER法适合用于表征EPS中的铀含量及形态。EPS中可溶性铀和颗粒铀的百分比,以及颗粒铀的粒径分布列于表1。EPS中可溶性铀的百分比在初始阶段(t=8 h)明显上升,然后随着铀固定反应的进行逐步下降,说明初始阶段EPS中主要发生铀酰离子的吸附,然后溶解态粒子逐步发生生物还原或生物矿化转化为铀颗粒。当反应进行到44 h时,颗粒铀占EPS中固定总铀量的66.6%,表明反应结束时颗粒铀是EPS中铀的主要形式。这一结果与William等[28]的研究结果一致,ShewanellaoneidensisMR-1的EPS与U(Ⅵ)反应形成了约3.0 nm的铀矿。CER法提取的EPS中颗粒铀较超声法和离心法多,进一步说明CER法对铀的萃取效率较高。
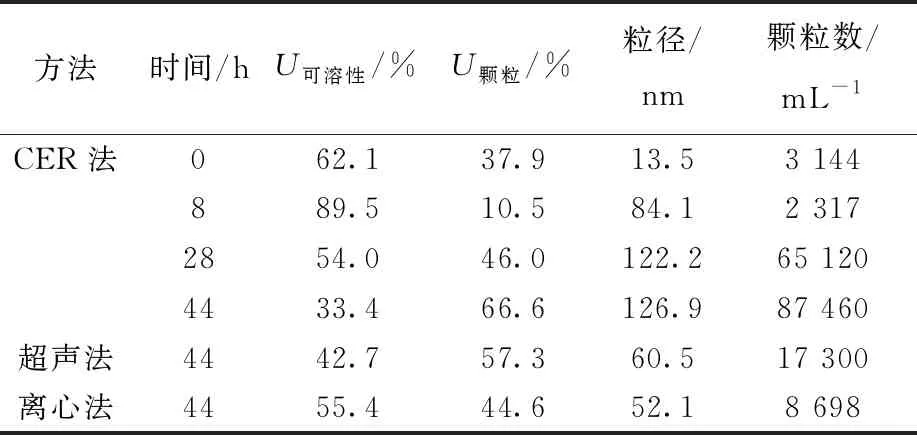
表1 EPS中可溶性铀和颗粒铀的百分比及颗粒铀的粒径Table 1 Fraction of soluble and particulate U and size of particulate U in EPS
CER法提取的EPS中铀颗粒的粒径分布示于图6。由图6可见,铀粒子随时间的推移逐渐形成铀颗粒并增大,颗粒数从3 144 mL-1增加到87 460 mL-1,平均粒径从13.5 nm增加到126.9 nm。在腐败希瓦氏菌固定U(Ⅵ)的初期,EPS中铀颗粒的数量减少,平均粒径增大,但EPS中铀含量略有增加,表明较小的铀粒子聚集并长大,在EPS中随时间积累。需要注意的是,ICP-MS得到的铀粒子的直径以铀原子计,不是含铀粒子的真实直径。然而由于铀原子远大于磷、氧等原子,所以该直径非常接近实际粒子的真实尺寸。

图6 CER法提取的EPS中铀颗粒的粒径分布Fig.6 Size distribution of nano-sized U particles in EPS extracts by CER method
上述结果表明,腐败希瓦氏菌的EPS在与U(Ⅵ)反应后,含有铀酰离子和铀矿物,说明EPS固定U(Ⅵ)过程中发生了生物吸附和生物矿化作用。
2.4 EPS对U(Ⅵ)的吸附机理


图7 EPS与铀作用前后的红外光谱Fig.7 FT-IR spectra of EPS before and after reaction with U(Ⅵ)
3 结论
1) 在处理过含铀地下水的腐败希瓦氏菌EPS中发现大量铀,占腐败希瓦氏菌固定铀总量的(14.0±1.0)%(CER提取法)。
2) SP-ICP-MS分析表明,EPS结合的铀有两种形态:溶解态和颗粒态。除生物吸附外,生物矿化也有助于腐败希瓦氏菌的EPS固定铀。
3) FT-IR分析结果说明,EPS吸附U(Ⅵ)时主要与EPS中的羧基官能团结合。
猜你喜欢
中国人兽共患病学报(2024年2期)2024-03-15 02:41:52
华声文萃(2022年11期)2022-06-10 05:13:44
文萃报·周五版(2022年43期)2022-05-30 10:48:04
当代水产(2022年1期)2022-04-26 14:35:38
实用老年医学(2021年12期)2021-12-05 22:16:09
黑龙江大学自然科学学报(2021年4期)2021-11-19 07:05:02
中国调味品(2017年2期)2017-03-20 16:18:21
现代检验医学杂志(2016年3期)2016-11-15 01:59:48
动物营养学报(2015年10期)2015-12-01 02:26:34
华东理工大学学报(自然科学版)(2014年5期)2014-02-27 13:49:27
