
肝X受体调控脂代谢研究进展
2021-07-30 02:15:52韩汶润张丹丹杜晓鹂董鹏志
中国药理学与毒理学杂志 2021年5期
韩汶润 ,张丹丹,朱 彦,杜晓鹂,董鹏志
(1.天津中医药大学组分中药国家重点实验室,天津 301608;2.天津国际生物医药联合研究院中药新药研发中心,天津 300457)
胆固醇是维持细胞膜流动性和产生各种激素及胆汁酸的重要脂质。因此,维持胆固醇稳态(包括吸收、输运、生物合成和外排)至关重要。胆固醇稳态的失调可能导致各种疾病,如动脉粥样硬化[1]等。肝X受体(liver X receptor,LXR)是胆固醇和脂质代谢的重要调节因子。在动物模型中,LXR活化可减缓心血管疾病并减少动脉粥样硬化的发生[2]。动脉粥样硬化发展中的一个关键事件是巨噬细胞募集到血管壁的内皮层及巨噬细胞对氧化和(或)修饰胆固醇的过量摄取。巨噬细胞不断积累氧化和(或)修饰的胆固醇及相关的炎症反应,导致泡沫细胞形成并促进动脉粥样硬化早期发展。逆转巨噬细胞胆固醇积累的过程和抑制血管壁炎症反应被认为是潜在的治疗动脉粥样硬化的新策略[3]。胆固醇逆转运(reverse cholesterol transport,RCT)可促进巨噬细胞内胆固醇清除,减少泡沫细胞形成,从而阻断动脉粥样硬化发生。LXR可促进RCT,对体内脂质代谢平衡的调节十分重要。LXR除具有改善脂代谢的作用以外,还参与调节多种炎症因子及炎症介质的表达,同时调节体内糖代谢过程,发挥抗动脉粥样硬化作用。LXR的多重脂调节功能揭示了开发LXR配体预防和治疗心血管疾病的广泛前景。但由于LXR激动剂易引发肝脂肪变性和高甘油三酯血症等不良反应而限制了其发展。因此,需要寻求新的防治策略,如开发LXRβ激动剂和组织特异性激动剂等。为此,本文就LXR信号通路涉及的分子及其在调控脂代谢中的作用予以综述,并对LXR的天然产物配体及合成配体进行简要总结。
1 肝X受体概述
1.1 结构
LXR是核受体家族成员之一,最早在1995年由Willy等[4]从肝cDNA文库中分离得到,因其在肝中表达丰富而得名。LXR具有典型的核受体结构,由4个结构域组成:①N端刺激转录的非配体依赖性功能活化域;②DNA结合域;③配体结合和受体二聚化所需的疏水配体结合域(ligand bound domain);④C端配体依赖性反式激活序列,也称为转录活化域[5](图1)。LXR包括LXRα和LXRβ 2种亚型,二者在DNA和配体结合域的氨基酸同源性约为77%[6]。LXRα主要在代谢活跃的肝、肾、小肠、脾及脂肪组织和巨噬细胞中高表达,其中在肝组织表达最高;而LXRβ在全身多种组织中均有表达[7]。
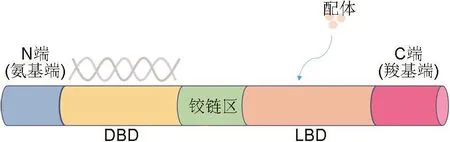
图1 肝X受体(LXR)的核受体结构.DBD:DNA结合域;LBD:配体结合域.
1.2 转录激活
LXR属于核受体Ⅱ类亚家族,需要与类视黄醇X受体(retinoid X receptor,RXR)形成LXR/RXR异二聚体才能发挥其调控功能。该异二聚体可被LXR与RXR的配体单独或共同激活,共同激活时起到协同诱导作用[8]。LXR/RXR异二聚体与靶基因启动子区域内的特定DNA序列即LXR反应元件(LXR response element,LXRE)结合,调节下游基因表达(图2)。LXRE是由4个核苷酸隔开的2个保守的六核苷酸序列(5′-AGGTCA-3′)以直接重复的方式排列,称为直接重复元件4(direct repeat 4)[6]。LXR的转录活性与LXR/RXR异二聚体密切相关,在无配体的情况下,LXR/RXR异二聚体与辅阻遏物结合时仍与目标基因的启动子区域结合,从而抑制了目标基因的激活[9]。当配体与LXR或RXR结合后,LXR/RXR异二聚体的构象发生改变,导致辅阻遏物如核辅阻遏物1(nuclear co-repressor 1)及视黄酸和甲状腺激素受体沉默介体(silencing mediator of retinoic acid and thyroid hormone receptor,也称为核辅阻遏物2)解离,并募集辅助激活因子,如受体辅助激活因子1(receptor co-activator 1)和激活信号辅助因子2(activating signal co-integrator 2),从而启动转录活性[10]。

图2 肝X受体的转录激活.DR4:直接反应元件4;LXRE:LXR反应元件;RXR:类视黄醇X受体.
2 肝X受体靶基因及其功能
LXR是调控体内胆固醇和脂质代谢平衡的重要转录因子。LXR主要通过几方面的协同作用调控胆固醇稳态:①肠道饮食中胆固醇的吸收;②肝胆固醇的生物合成和摄取;③高密度脂蛋白(high density lipoprotein,HDL)颗粒的合成和重塑及随后的逆向胆固醇转运;④肝中将胆固醇转化为胆汁酸并排泄到胆汁中[11]。此外,LXR与脂肪酸生物合成及外周血中甘油三酯水平相关。
LXR的2种亚型在体内甾醇稳态中发挥独特作用。在低密度脂蛋白受体(low-density lipoprotein receptor,LDLR)-/-小鼠背景下,LXRα-/-和LXR β-/-单基因或双基因敲除实验表明,LXRα和LXRβ均有抗动脉粥样硬化作用,但LXRβ贡献弱于LXRα,且2种亚型的功能有所区别,即LXRα在激动剂作用下启动转录,而LXRβ可能在配体存在时抑制转录[12]。
2.1 调控胆固醇逆转运
LXR可刺激胆固醇从外周细胞排出到肝,然后通过胆汁酸分泌,最终通过粪便排出体外,这一过程被称为RCT。LXR被激活后,可促进肝细胞及巨噬细胞的胆固醇外流,并通过细胞色素P450 7A1(cytochrome P450 7A1,CYP7A1)将游离胆固醇转化为胆汁酸,或经肠胆固醇排泄清除含载脂蛋白B(apolipoprotein B,ApoB)的脂蛋白胆固醇。此外,LXR激活并诱导脂肪生成基因固醇调节元件结合蛋白1c(sterol regulatory element-binding protein 1c,SREBP1c)、脂肪酸合成酶(fatty acid synthase,FAS)、硬脂酰辅酶A去饱和酶1(stearoyl-coenzyme A desaturase 1,SCD1)和LDLR诱导型降解子(inducible degrader of the LDLR,IDOL)的表达(图3)[13-14]。其中,依赖ATP结合盒转运蛋白 A1(ATP-binding cassette transporter A1,ABCA1)的胆固醇通过HDL从巨噬细胞外排的过程,在人心血管疾病的生理和病理进程中发挥重要作用[15]。下面将简要介绍LXR靶基因的生理功能。

图3 肝X受体调控胆固醇逆转运.方框内所示为LXR靶基因,可被LXR激动剂调控.ABCA1:ATP结合盒转运蛋白A1;SR-B1:清道夫受体B1;LXRα:肝X受体α亚型;SREBP1c:固醇调节元件结合蛋白1c;ChREBP:糖反应元件结合蛋白;SCD1:硬脂酰辅酶A去饱和酶1;FAS:脂肪酸合酶;CYP7A1:细胞色素P450 7A1(也称为胆固醇-7α-羟化酶);TICE:经肠胆固醇排泄;IDOL:LDLR诱导型降解子;LDLR:低密度脂蛋白受体;LDL:低密度脂蛋白;HDL:高密度脂蛋白;Pre-β HDL:HDL前体;VLDL:极低密度脂蛋白;CE:胆固醇酯;CETP:胆固醇酯转运蛋白;TG:甘油三酯;NPC1L1:类尼曼-匹克C1型蛋白;LPL:脂蛋白酯酶.
2.1.1 三磷酸腺苷结合盒转运体家族
目前,已知的胆固醇外流途径有:①游离胆固醇分子通过水相扩散结合到HDL颗粒;②清道夫受体B1(scavenger receptor class B type 1,SR-B1)介导途径;③ABCA1介导磷脂和胆固醇从细胞向低脂Apo如ApoA1的主动外排途径。而ABCA1参与的胆固醇外流是巨噬细胞RCT的主要途径[16],这一过程促使细胞磷脂分子从细胞质小叶转移到细胞质膜的外表面小体并促进ApoA1结合到细胞表面[17]。ABCA1广泛参与肠、肝和巨噬细胞中的胆固醇和磷脂转运,其功能缺失会导致严重后果。如在丹吉尔病(Tangier disease)中,ABCA1突变会阻断胆固醇和磷脂在肠上皮细胞和肝细胞基底外侧膜的转运,致使因缺乏足够的胆固醇和磷脂而影响ApoA1蛋白脂化。这些空载的ApoA1则被肾迅速清除出循环系统,进而导致严重的HDL缺乏综合征[18]。
ABCG1也参与调控细胞内胆固醇稳态。生理条件下,ABCG1的受体包括较大HDL2颗粒(极大HDL和大HDL)与较小HDL3颗粒(中型HDL、小型HDL和极小型HDL)[19]。研究发现一种独特现象,即ABCA1和ABCG1在刺激胆固醇外排和抑制动脉粥样硬化方面发挥着协同作用[20]。一方面,ABCA1介导磷脂和胆固醇向低脂或无脂ApoA1的转移,以生成新生的HDL颗粒;另一方面,上调ABCG1表达可促进胆固醇向HDL的流出。
而ABCG5和ABCG8也是LXR的直接靶基因,通常形成异二聚体ABCG5/ABCG8发挥作用。它们在内质网二聚化后,输运至肠细胞的顶膜和肝细胞的小管膜,将细胞内固醇泵入肠腔和胆汁中,并最终通过粪便排出体外[21]。
2.1.2 载脂蛋白E
ApoE作为脂蛋白的组成部分,在维持血浆脂蛋白稳态中起着关键作用。ApoE主要由肝合成,其他器官和细胞,如脑、肾、脂肪细胞、平滑肌细胞和巨噬细胞也能产生ApoE[22]。ApoE主要通过ABCA1进行脂质化,而二者均受LXR的转录调控[23]。
2.1.3 胆固醇酯转移蛋白
胆固醇转移到HDL颗粒后,经卵磷脂胆固醇酰基转移酶酯化形成胆固醇酯,最终形成成熟的HDL。肝脂肪酶和内皮脂肪酶分别介导甘油三酯和磷脂水解进而重塑HDL颗粒[24]。在表达胆固醇酯转运蛋白的物种中,大部分HDL胆固醇酯可通过胆固醇酯转运蛋白转移至含ApoB的脂蛋白中,这些脂蛋白由肝细胞表面LDLR摄取从而促进RCT在体内运行[25]。
2.1.4 脂质转运蛋白
磷脂转运蛋白参与低脂HDL前体(pre-β HDL)的生成,为细胞胆固醇提供更多的受体,并参与维持循环系统中HDL的含量稳定[26]。脂蛋白脂酶是脂蛋白代谢的关键酶,专司循环脂蛋白中甘油三酯的水解,向外周组织释放游离脂肪酸。在肝组织中,脂蛋白脂酶能促进HDL胆固醇的摄取,从而促进胆固醇的逆向转运[27]。
2.1.5 CYP7A1
CYP7A1是胆固醇到胆汁酸的经典转化途径中的限速酶[28]。CYP7A1属于细胞色素P450蛋白家族。与人不同,啮齿类动物在CYP7A1基因启动子中含有LXRE序列。此外,LXRα(而非LXRβ)是小鼠肝CYP7A1 mRNA表达的主要调节因子[29]。除胆汁酸的合成代谢外,LXR也参与调节胆汁酸的分解代谢。有研究表明,配体激活的LXRα通过与启动子中的LXRE序列结合而上调人UDP葡萄糖醛酸转移酶1家族多肽A3(UDP glucuronosyltransferase 1 family polypeptide A3)(葡糖醛酸结合胆汁酸最活跃的酶之一)基因的表达,从而使胆汁酸葡萄糖醛酸化,并最终转化为可通过尿液排泄的代谢产物[30]。
2.2 调控胆固醇合成、分解和代谢
胆固醇的积累和清除分别由2种转录因子调节。在低胆固醇条件下,SREBP2促进胆固醇的合成和摄取,而细胞胆固醇水平过高时,LXR则促进胆固醇外流。研究表明,SREBP2作为ABCA1基因表达的正调节因子,通过诱导下游基因生成LXR的氧甾醇配体发挥作用[31]。而LXR则通过肝表达LXR诱导序列(liver-expressed LXR-induced sequence,LeXis)(一种非编码 RNA链)[32]和内质网膜泛素连接酶环指蛋白145(ring finger protein 145,RNF145)[33]抑制胆固醇的生物合成。其主要机制是LeXis与异质核核糖核蛋白(RALY heterogeneous nuclear ribonucleoprotein)相互作用并影响后者与基因组DNA的相互作用[32]。RNF145诱导SREBP蛋白裂解激活蛋白将靠近外被体蛋白复合物Ⅱ(coat protein complex Ⅱ)结合位点的2个赖氨酸残基泛素化,进而抑制其向高尔基体的转运和SREBP2的后续加工[33]。
LXR还调节细胞内胆固醇的积累。研究发现,LXR通过调控IDOL抑制LDLR通路。已知IDOL是一种E3泛素连接酶,可触发LDLR在细胞质内泛素化而被靶向降解[34]。LXR-IDOL途径为SREBP2途径提供了补充,即SREBP2途径在低胆固醇条件下增加LDLR转录,而增加LDL胆固醇的摄取[35]。
类尼曼-匹克C1型蛋白1(Niemann-Pick C1-like1,NPC1L1)分布于肠上皮细胞刷状缘膜,其功能包括介导肠细胞摄取游离胆固醇、将新分泌的胆汁胆固醇转运回肝细胞,从而防止内源性胆固醇的过度流失。LXR可调控NPC1L1在小鼠和人肠细胞中的表达,降低肠道胆固醇的吸收[36]。
2.3 调控脂肪酸代谢
LXR激活后,通过诱导SREBP1c、糖反应元件结合蛋白(葡萄糖敏感转录因子,可将肝中过剩的糖类转化为脂质)及SREBP1c靶蛋白FAS和SCD1的表达,促进脂肪酸的生物合成[13]。在脂肪组织中,LXR调节脂结合蛋白和代谢蛋白如ApoD和甲状腺激素应答基因的表达,并可能通过诱导脂肪酸在线粒体中的β-氧化而促进其分解。此外,LXR通过诱导葡萄糖转运蛋白4促进葡萄糖摄取。综上,LXR影响脂肪组织中碳水化合物和脂质代谢,促进葡萄糖摄取,增强脂肪酸β-氧化及其分解[37]。
3 调控炎症反应
除调节脂质稳态外,LXR同时参与调控多个炎症信号通路。例如,LXR对白细胞介素18(interleukin-18,IL-18)和IL-1β的调节具有多重作用。LXR抑制脂多糖诱导的骨髓源性巨噬细胞IL-1β和IL-18表达水平,同时负向调节IL-18前体胱天蛋白酶1的水平,从而抑制IL-18的成熟,且LXR激活后通过调节干扰素调节因子8增强IL-18BP(IL-18的有效内源性抑制剂)的表达[38]。配体激活的LXR可以抑制典型炎症基因的表达(如诱导型一氧化氮合酶、环氧合酶2和基质金属蛋白酶9)和各种趋化因子对脂多糖、肿瘤坏死因子α和IL-1β刺激的反应[39]。
有报道称,LXR通过以小泛素相关样修饰物(small ubiquitin-like modifier,SUMO)信号通路依赖的方式与NF-κB转录复合物相互作用来抑制炎性基因表达[40]。而Ito等[41]报道,在无SUMO的条件下,LXR激活仍发挥抗炎作用,其机制主要是通过调控固醇转运蛋白ABCA1的转录,改变膜胆固醇稳态,抑制Toll样受体下游的NF-κB和丝裂原活化蛋白激酶介导的炎症信号。
Kappus等[42]发现,在缺乏ABCA1 和ABCG1的条件下,激活LXR仍产生有效的抗动脉粥样硬化作用,即LXR发挥抗炎作用不依赖于巨噬细胞ABCA1和ABCG1介导的胆固醇外流通路。而Westerterp等[43]研究表明,ABCA1和ABCG1缺陷导致骨髓细胞中的胆固醇积累并激活NOD样受体蛋白3炎性小体,促进动脉粥样硬化病变中的中性粒细胞浸润和网状病变。另有研究发现,LXR激动剂通过顺式抑制(cis-repression)LXR和调节胆固醇代谢可抑制体内无菌性炎症时中性粒细胞的迁移[44]。综上,LXR通过多种途径调节炎症信号通路,其具体机制需要深入研究。
4 肝X受体配体
动脉粥样硬化的发展可导致心脏病发作和卒中等严重后果。LXR激活可发挥几种不同的抗动脉粥样硬化作用,如增加外周胆固醇的流出、促进胆固醇向肝的逆向转运及胆汁酸分泌、抑制炎症、提高巨噬细胞的存活率并减少其对含ApoB脂蛋白的摄取[45],充分表明LXR是治疗动脉粥样硬化的潜在药物靶标。综上,通过配体激活LXR控制胆固醇通量从而预防和治疗心血管疾病具有很好的应用前景。
LXR的整体结构尤其是配体结合口袋(ligandbinding pocket)可非常灵活地与结构高度不同的配体结合。激动剂在配体结合口袋中的结合导致LXR-RXR异二聚体的受体构象发生变化,从而影响异二聚体的转录活性(图4)[46]。

图4 肝X受体/类视黄醇X受体异二聚体整体结构示意图.A和B:RXRα/LXRβ沿DNA方向(5′到3′)结构和侧面结构(蓝色:LXRβ;品红色:RXRα;青色:配体;灰色:辅因子肽);C:RXRα/LXRβ异二聚体中结构域位置;D:LXRα/RXRβ异二聚体LBD的结构(黄色:LXRα-LBD;紫色:RXRβ-LBD;绿色:LXR羧端配体依赖的转录活化域螺旋).
4.1 内源性配体
4.1.1 内源性激动剂
最初研究认为,LXR是一种孤儿受体。但后续研究证实,氧化甾醇是LXR的内源性配体。氧化甾醇是胆固醇的氧化衍生物,包括24(S)-和25-环氧胆固醇及22(R)-、24(S)-、20(S)-和 27-羟基胆固醇(但不包括胆固醇)均是LXR的配体。22(R)-和20(S)-羟基胆固醇是甾体激素合成过程中的中间产物。24(S)-羟基胆固醇在大脑中产生,是循环系统中最丰富的氧化甾醇,而24(S)-和25-环氧胆固醇主要存在于肝中[47]。
4.1.2 内源性拮抗剂
花生四烯酸和其他不饱和脂肪酸竞争性地阻断LXR的激活,抑制T0901317诱导的内源性SREBP1c基因表达升高。前列腺素F2α(花生四烯酸的环氧合酶代谢产物之一)以剂量依赖性方式拮抗T0901317诱导的LXRα-LBD和LXRβ-LBD激活,并拮抗ABCA1和ABCG1启动子的激活[48]。
4.2 天然产物配体
LXR的配体包括很多天然产物或其衍生物(表1)。研究发现,花青素可与LXRα和LXRβ结合,调控靶基因如ABCA1和ABCG5的表达,但对LXRα的亲和力高于 LXRβ(LXRα EC50为 3.5 μmol·L-1,LXRβ为125.2 μmol·L-1)。单萜桉树脑是在茶和草药精油中发现的成分,可组织特异性激活LXRα和LXRβ。桉树脑(50~200 μmol·L-1)处理RAW264.7巨噬细胞后,ABCA1和ABCG1 mRNA水平显著升高;桉树脑未显著影响HepG2肝细胞中SREBP1c mRNA表达,而FAS和SCD1 mRNA表达显著下降。芍药苷是一种单萜苷,用于治疗高脂血症和高血糖。荧光素酶报告基因检测发现,芍药苷10 μmol·L-1可显著激活HepG2细胞LXRα。槲皮素或槲皮素糖苷被胃肠道吸收后,形成槲皮素-3-O-葡萄糖苷。槲皮素-3-O-葡萄糖苷(50 μmol·L-1)可增加RAW264.7巨噬细胞中ABCA1 mRNA和蛋白水平[49]。
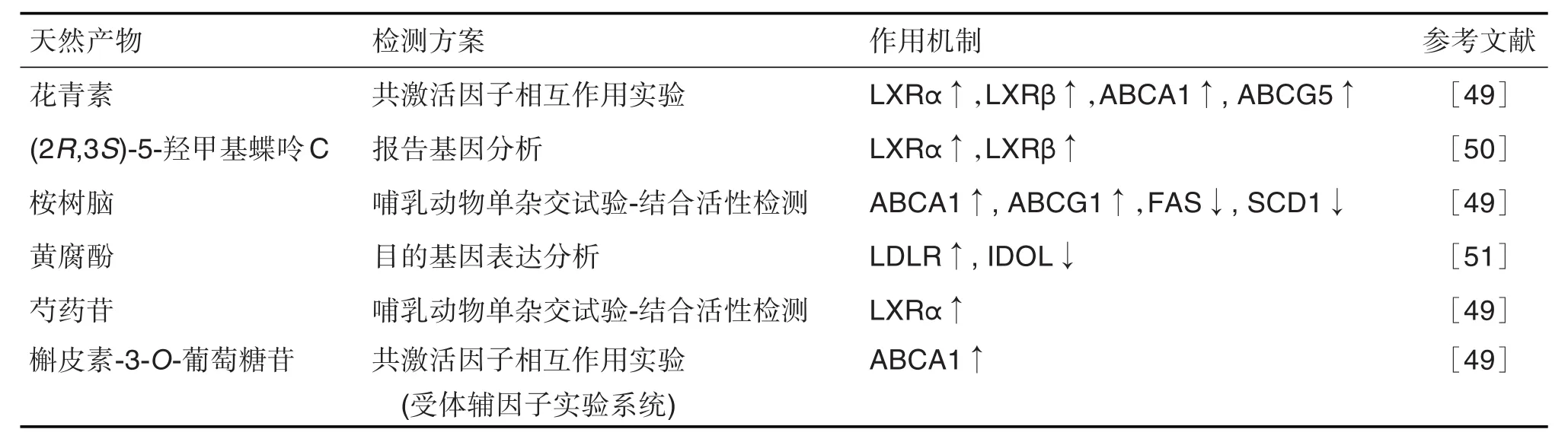
表1 肝X受体的天然产物配体
从植物气生部分分离出的化合物,如蝶呤倍半萜(2R,3S)-5-羟甲基蝶呤C,亦可激活LXRα和LXRβ,比 GW3965(10 μmol·L-1)对 2 种 LXR 亚型的激活更明显[50]。Chen等[51]研究发现,黄腐酚可增加细胞表面LDLR表达,进而增加HepG2细胞中LDL摄取;黄腐酚(10和20 μmol·L-1)可显著降低IDOL mRNA和蛋白的表达。此外,与T0901317单一处理相比,用黄腐酚 20 μmol·L-1预处理 HepG2细胞后与T0901317共孵育,IDOL mRNA和蛋白质水平均降低,LXRα mRNA和蛋白水平无明显改变,表明黄腐酚可抵消LXR的激活。
4.3 合成配体
经典LXR合成类激动剂有T0901317和GW3965等[52]。在非临床动物模型中,合成的LXR激动剂可改善动脉粥样硬化和广泛的炎症性疾病,但系统性LXR激活可引发肝脂肪变性和高甘油三酯血症(LXRα在肝中诱导脂肪生成基因表达的后果)。近来研究出一些新的防治策略,包括LXRβ选择性激动剂、组织选择性LXR激动剂和转抑制选择性LXR激动剂等[14]。不同种类的LXR合成配体见表2。
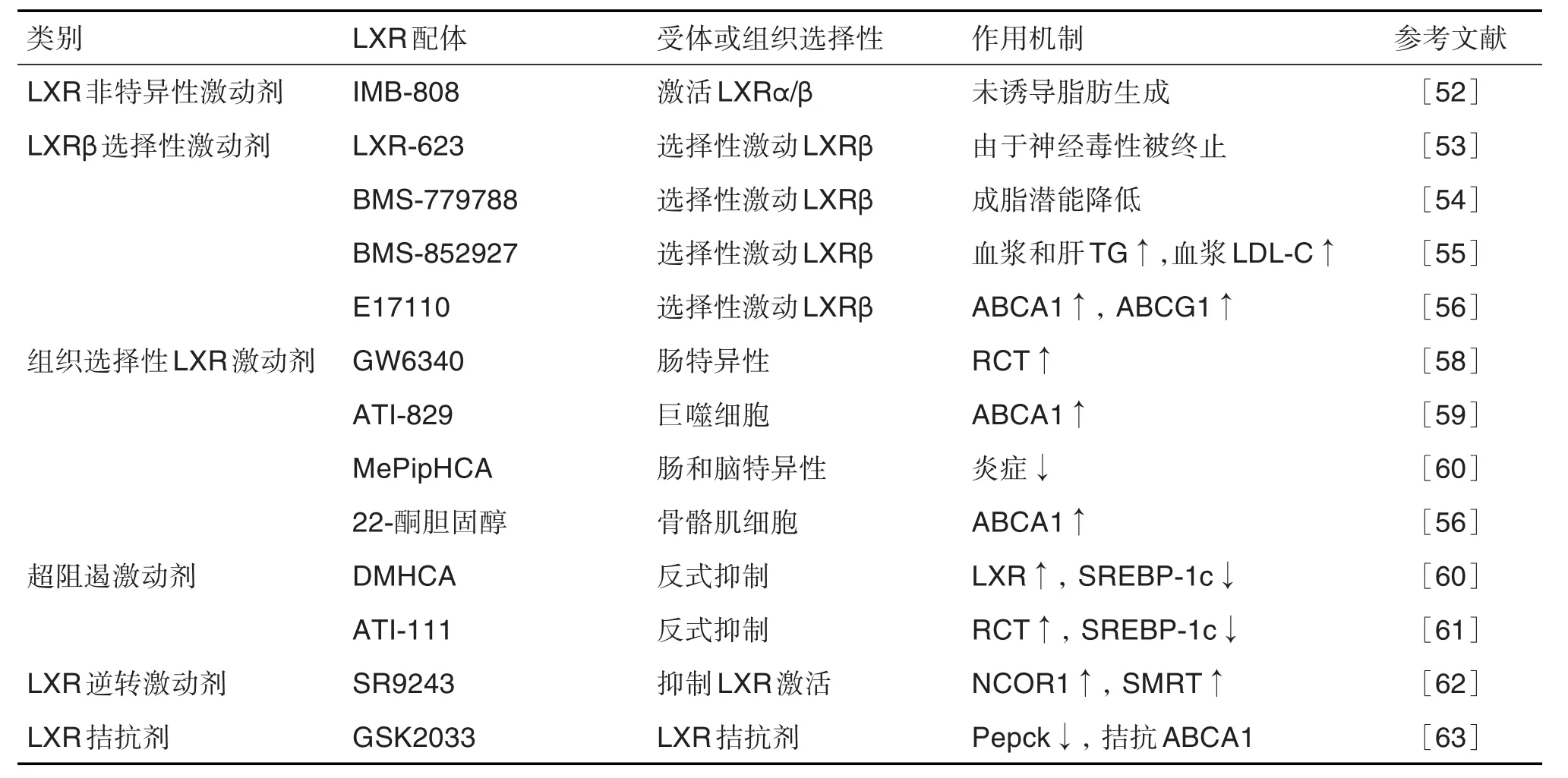
表2 肝X受体的合成配体(激动剂和拮抗剂)
4.3.1 非特异性激动剂
IMB-808激活LXRα和LXRβ,在多个细胞系中有效增加与RCT以及与胆固醇代谢途径有关基因的表达,尤其显著促进RAW264.7和THP-1巨噬细胞的胆固醇外流,并相应减少细胞脂质的积累,且不会上调脂肪生成基因的表达[52]。
4.3.2 肝X受体 β选择性激动剂
LXRβ激动剂LXR623可增加外周血细胞中LXR靶基因ABCA1和ABCG1的表达,但最高剂量时可引起中枢神经系统和精神疾病等不良反应,导致其研发终止[53]。BMS-779788和BMS-852927在食蟹猴或小鼠中选择性激活LXRβ,可显著改善血脂异常,诱导RCT相关基因表达且具有较低的成脂潜能[54-55]。与其他LXR激动剂相同,LXRβ激动剂E17110在体外可增加ABCA1和ABCG1表达,并增强RAW264.7巨噬细胞中的胆固醇外流,表明其可能具有抗动脉粥样硬化活性[56]。
4.3.3 组织选择性激动剂
Li等[57]建立了62个甾醇衍生物库并进一步设计、合成和测试了12个甾醇衍生物的生物学性质,证明化合物4和6是LXR激动剂,可以抑制巨噬细胞泡沫细胞的形成而不会诱导肝细胞或脂肪细胞中甘油三酯的累积。GW6340是肠特异性激动剂,显著上调小肠中的ABCA1,ABCG5和ABCG8的表达,促进巨噬细胞RCT,但在肝中无此作用[58]。ATI-829可在THP-1巨噬细胞中强烈诱导ABCA1表达,且在HepG2肝癌细胞中未明显上调SREBP1c表达。LDLR-/-小鼠给药后,ATI-829激活了肠道和巨噬细胞中的LXR靶基因,但未激活肝中脂肪酸合成基因而未增加肝和量[59]。MePipHCA是一种基于甾醇的LXR激动剂,抑制葡聚糖硫酸钠结肠炎和创伤性脑损伤2种模型小鼠炎症而未引起肝脂质蓄积或肝损伤[60]。
4.3.4 其他
最近开发的LXR激动剂还有反向激动剂等类型[62]。Zhang 等[64]报道,含有合成 LXR 激动剂GW3965的纳米颗粒〔(乳酸-共聚乙二醇)-b聚(乙二醇)共聚物〕具有抗炎作用,并在不引起肝脂肪变性的情况下抑制动脉粥样硬化的发展。
5 结语
LXR在调控胆固醇代谢过程中起到核心作用。此外,LXR在调控糖代谢、脂质代谢、炎症反应以及先天免疫等方面也发挥重要作用。随着对LXR在动脉粥样硬化疾病发展中的作用及合成LXR配体的深入研究,以LXR为靶点寻找激动剂,在治疗高脂血症和动脉粥样硬化等疾病方面将具有很好的应用前景。LXR协调抑制炎症的能力对于理解脂质代谢和炎症之间的相互作用以及药物发现具有重要意义,但LXR的这种协调作用仍然存在很多疑问。目前常用的LXR合成激动剂,多会引起脂肪在肝的积累以及血中甘油三酯水平升高。LXR的功能研究为动脉粥样硬化和众多炎症性疾病的治疗提供了潜在的机会。然而,由于啮齿类动物以及非人类灵长类动物与人之间的种属差异,难以预测LXR激动剂对人体的不良反应,致使LXR激动剂的研究进一步复杂化且困难重重。因此,深入研究LXR的作用机制,充分了解LXR调控脂质代谢的组织特异性,将有助于开发更加安全有效的LXR选择性激动剂,也将为开发新型的抗动脉粥样硬化药物及多种心血管疾病的治疗提供理论基础。此外,将LXR激动剂与其他可减轻脂肪生成的药物(如IDOL拮抗剂)进行联合用药也是一种可行方案。随着LXR激动剂作用机制研究的不断深入,综合结构信息指导下的药物研发,最终会实现靶向药物的有效和安全性应用。
猜你喜欢
中成药(2018年10期)2018-10-26 03:41:22
中成药(2018年9期)2018-10-09 07:18:36
天然产物研究与开发(2018年6期)2018-07-09 06:01:46
中成药(2018年1期)2018-02-02 07:19:53
中成药(2017年4期)2017-05-17 06:09:26
材料科学与工程学报(2016年4期)2017-01-15 13:35:48
合成化学(2015年4期)2016-01-17 09:01:11
医学研究杂志(2015年5期)2015-06-10 06:43:26
中成药(2014年9期)2014-02-28 22:28:50
无机化学学报(2014年6期)2014-02-28 17:32:06
