
数学模型预测药源性肝损伤研究进展
2021-07-30 02:15:52李思泽相小强
中国药理学与毒理学杂志 2021年5期
李 敏,李思泽,姚 莉,相小强
(复旦大学药学院,上海 201203)
药源性肝损伤(drug-induced liver injury,DILI)是一种常见的药物不良反应,严重时可造成患者急性肝衰竭和死亡。DILI是近年来上市药物被召回的重要原因之一,也是药物标签更改、增加黑框警告的常见原因之一。目前市场上仍有>1000种药物具有肝毒性风险[1]。DILI已成为药物开发、监管和使用人员所关心的重要安全性问题。
目前,全球DILI的发生率约为19/100 000[2],我国总人口中的发生率约为23.8/100 000[3],高于全球水平。主要原因在于我国人口基数大、临床药物种类多(包含西药和中药等)、不规范用药情况普遍及医务人员和公众对DILI的认知不足等[4-5]。而且我国DILI患者中有87%为急性DILI,容易发展为急性肝衰竭。但目前DILI治疗方法并不多,仅能及时发现并停用可疑药物,给予保肝和抗炎治疗[5-6]。
虽然大部分患者治愈及好转率>90%[5],但DILI尚存在难以及时诊断和鉴别的问题,且DILI的治疗滞后易为患者带来很大危害。为避免上市药物发生肝毒性反应,药物肝毒性已成为药物研发和临床应用过程中倍受关注的重要安全性问题。在药物研发阶段,药物是否具有肝毒性及肝毒性程度可通过动物实验和临床试验进行判断或预测,其中动物实验一般以实验动物的肝病理变化作为判断指标,而临床试验则一般以血清谷丙转氨酶(glutamic-pyruvic transaminase,GPT)或谷草转氨酶(glutamic-oxaloacetic transaminase,GOT)≥3倍正常值上限,作为肝损伤发生的早期敏感信号和判断标准[5-7]。
由于动物与人体对药物毒性具有一定的种属差异,动物实验结果往往不能反映药物对人体的毒性。Fasiglifam(TAK-875)是一种治疗糖尿病的候选药物,啮齿动物的毒性实验结果表明,该药不会诱发动物肝损伤,但在进入临床Ⅲ期试验后发现,治疗组出现GPT≥3倍正常值上限的概率比安慰剂组高,且剂量越大产生肝毒性的概率越高,因此该药的研发被中断[8]。临床试验中一旦出现肝损伤信号,该药物的临床试验即需停止,这就意味着药物研发的前期努力付之东流。药物即使能够成功上市,并不代表该药的肝毒性监管就此结束。曲格列酮在被批准用于治疗2型糖尿病后不久,即因其肝毒性而退出市场。但在体外毒性试验、动物实验和临床试验中,均未发现其具有肝毒性,主要原因在于此诊断肝损伤的标准仍具有局限性[9]。
虽然生物标志物(如GPT)已经是DILI早期预警信号,但仍不能在药物研发过程中对DILI进行早期预测和诊断。由于动物研究伦理要求变高且效率低,依靠动物实验判断药物肝毒性的方法不再成为DILI研究的首选。同时,不同种属间的肝毒性差异也为DILI的准确判断带来许多困难。因此,欧盟与美国出台了一系列政策鼓励以非动物实验作为评价药物安全性的方法[10]。众多研究者将药物肝毒性的判断和预测从传统的动物实验,转向基于细胞或细胞器的分子生物学实验和基于数学模型的非生物学实验的替代方法。基于数学模型的肝毒性预测方法,主要包括基于化合物结构定量构效关系(quantitative structure-activity relationship,QSAR)模型、基于毒理基因组学模型和基于人生理药动学(physiologically based pharmacokinetic,PBPK)模型的方法。不少以QSAR模型为基础的计算工具和软件已经上市应用,如Toxmatch®,Toxtree和ADMET predictor®等。以PBPK模型为基础的肝毒性预测工具如DILIsym®,也已经开始应用。
本文介绍目前3类基于数学模型预测药物肝毒性的方法——基于QSAR模型、毒理基因组学模型和PBPK模型的方法,以期为DILI的早期预测和肝毒性药物临床合理应用提供参考。
1 基于定量构效关系模型的方法
QSAR模型使用数学模型描述化合物结构与其性质之间的关系[11],根据已知化合物的结构信息推测化合物的特定理化或生物学性质,可以对未知化合物性质进行定性或定量预测[12]。目前,使用QSAR模型预测肝毒性仍属于定性预测[13],主要分为4个步骤:①筛选肝毒性和非肝毒性化合物;②依据化合物结构计算并筛选分子描述符或指纹,构建学习集;③利用数学工具探寻定量-构效关系;④验证结果并优化模型[14-15]。
以Huang等[16]研究为例,他们选择了136种肝毒性和65种非肝毒性药物,根据化合物结构计算并筛选出1064个合适的分子描述符或指纹,使用随机森林(random forest,RF)算法构建模型,并使用10倍嵌套-交叉验证方法优化模型。预测结果发现,最佳模型的准确度最高为79.1%,验证集测试准确度为87.0%。也有研究者根据化合物肝毒性程度分别建立二分类模型(2-class model,包括DILI和no-DILI药物,即肝毒性药物和非肝毒性药物)和三分类模型(3-class model,包括 most-DILI、less-DILI和no-DILI药物,即强肝毒性药物、弱肝毒性药物和非肝毒性药物),并对比2种分类模型预测肝毒性结果的准确度。结果发现,三分类模型不仅预测结果准确度较高,而且具有更高的肝毒性分辨率,可以更好的区分非肝毒性和肝毒性药物[17]。为了提高肝毒性分类预测的分辨率,Liu等[18]也根据多种不良肝反应(adverse hepatic effects,AHE)与机器学习相结合,构建了1个三级肝毒性预测系统。该系统使用化合物结构作为基础数据,化合物肝毒性作为预测终点指标,4个肝毒性严重度和特定AHE作为分类指标。在收集2017种化合物和403种AHE相关的化合物信息后,使用RF算法构建了27个模型,获得了67.0%~78.2%的准确度。这类肝毒性分层预测系统为多角度分析候选药物性质提供了帮助。
目前,这类只使用单个机器学习算法的QSAR模型研究比较广泛,但由于所选数据的来源不同、内容不均衡和训练集化合物数量的差异,这类方法的精确度和稳定性变化较大,准确度在60%~90%不等[11],而且每一种建模算法的优缺点不同,在建模过程中可能会出现欠拟合和过拟合等问题,影响预测结果的准确性和可信度。
为扬长避短,也有研究者尝试使用多种机器学习算法,构建集成且预测效果更好的QSAR模型。集成模型预测结果更准确,且可避免单一模型的缺点[19]。集成模型的构建步骤与单个模型相似,但研究者会根据单一算法建模结果,构建含有不同模型的多个集成模型,并比较不同集成模型的预测结果,以获得一个最优的集成模型[20]。如Ai等[20]设计了一种集成模型预测DILI的方法,从文献中筛选并建立了1个含1241种化合物的分子描述符数据库后,同时使用3种算法:支持向量机(support vector machine,SVM)、RF和极端梯度增强(extreme gradient boosting,XGBoost)算法及 12 个分子指纹,建立了36个基础算法模型和35个集成模型,并使用5倍-交叉验证方法进行验证。结果显示,集成模型的预测效果普遍比传统单一模型更好,最佳集成模型是由5个分类器组成的,称为集成Top-5的模型,准确度为(71.1±2.6)%,验证集准确度为84.3%。而36个基础算法模型预测结果的准确度高低不齐,范围在62.7%~70.2%。
总体而言,基于QSAR模型预测肝毒性的方法的优势在于快速且高效,适用于药物研发前期对化合物进行高通量筛选,这有助于降低药物研发成本,缩短研发周期。目前也已有相关软件上市应用,如美国Simulation Plus公司所研发的ADMET Predictor软件和欧洲化学局联合中心(Joint Research Centre of European Chemicals Bureau)开发的免费毒性预测平台Toxtree。但基于QSAR模型预测肝毒性的方法的缺点也在于只依赖化学结构,且预测结果的准确性很大程度上取决于建模所选化合物的数量和种类,与各种建模算法无显著关系。化合物结构能反映化合物的一些理化性质和药效,但并不能完全表明化合物是否具有肝毒性。药物在人体是否引起DILI,不仅取决于化合物的结构,也与其在体内的暴露量有密切关系。而基于QSAR模型的方法忽略了药物在人体内的暴露量。如托卡朋(tolcapone)和恩他卡朋(entacapone)2种药物的结构类似,使用QSAR模型分析发现两者结构并无明显差异,但是前者由于具有肝毒性且可造成急性肝衰竭而退市,而后者却无肝毒性[21]。
2 基于毒理基因组学模型的方法
药物的毒性效应与基因表达之间存在着密切的联系,通过测试药物肝毒性所诱导的基因变化,即可探究某基因与药物肝毒性之间的关系。目前已有公开2个大规模毒理基因组学数据集:日本毒理基因组学数据库(Toxicogenomics Project-Genomics Assisted Toxicity Evaluation System,TG-GATEs)[22]和 DrugMatrix 数据库[23],这 2 个数据集可能为开发DILI预测模型带来新的进展[24]。
目前有研究者开始利用毒性基因组学数据,构建数学模型预测药物肝毒性的方法,根据体外实验和生物学分析确定肝毒性相关基因,并构建数学模型模拟该基因表达水平与药物肝毒性之间的定量关系,相关性越高证明肝毒性可能性越大。Zidek等[25]已预测出64个肝毒性的关键基因。为提高预测准确度,Su等[26]依据SVM递归特征消除(SVM-recursive feature elimination)、最小冗余最大相关性(min-redundancy max-relevance)和交叉验证创建了一种模型,并命名为MEMO,用于构建数学模型预测肝毒性。他们首先进行体外细胞实验,并获得多剂量下药物浓度信息,建立药物多剂量-反应曲线;随后根据体外实验结果使用MEMO方法筛选相关基因,并进行生物学分析,确定了10个肝毒性相关基因;最后使用TG-GATEs数据库信息进行了验证。实验结果表明,使用该预测模型可以准确高效地预测药物引起的肝毒性,准确度达到97%。与其类似,Cha等[27]选取了8种肝毒性药物和8种非肝毒性药物,使用药物的HepG2细胞毒性基因组学数据,建立了模型预测非甾体类抗炎药引起DILI的风险,模型确定了77个毒性相关基因,并成功对训练集外的4种药物(酮洛芬、萘丁美酮、吲哚美辛和尼氟酸)肝毒性进行了预测。同时有研究者发现,基于毒理基因组学数据预测肝毒性的结果要明显优于QSAR模型,前者正确分类率为76%,而后者只有61%[28]。但也有研究表明,毒性基因作为生物标志物预测肝毒性的效果,可能不如临床研究中所使用的生物标志物敏感[10]。基于毒物基因组学的预测模型并无预想的有效,部分原因是开发成本高,且缺乏先进的数据挖掘工具。另外,也可能在于基因表达改变并不足以诱发药物产生肝毒性变化,具体原因仍需进一步研究。
因此,预测肝毒性时,除考虑药物理化性质、肝毒性敏感性和毒性基因的变化,更需要考虑药物在体内浓度变化,尤其是在肝部位的暴露量,而这往往需要用到更先进的数学模型,如PBPK模型。
3 基于生理药动学模型的方法
DILI的发生一方面取决于肝细胞对药物或其代谢产物的敏感性,另一方面也取决于药物在肝的暴露浓度和持续时间。前者可通过肝细胞体外实验数据结合数学模型得到,而后者则需要借助PBPK模型。
PBPK模型是一种基于机体生理学知识,利用数学模型,对人体或动物体内的药动学特征——吸收、分布、代谢和排泄进行定量模拟和预测的方法[29-30]。PBPK模型根据解剖结构,将机体分为一系列的房室,每个房室代表某个器官组织或其一部分,并相应整合了这些器官的生理生化参数,包括器官体积、血流量、组织成分、细胞内外液比例、代谢酶和转运体表达量等。药物在各个房室之间的转移按照机体循环系统的流向进行,并遵循物料平衡原理。PBPK技术的一大优点是其强大的外推功能。例如,动物PBPK模型可以外推到人[31-33]。基于PBPK模型的方法可推广预测候选药物的肝毒性、药物肝毒性浓度和临床安全剂量范围[34],既可应用于药物非临床研究阶段,也可应用于临床试验阶段,甚至可以替代部分临床试验。而且,随着数学模型的发展,越来越多的模型被应用到肝毒性的预测中。根据数学模型的不同,基于PBPK模型预测肝毒性的方法可以分为以下几类。
3.1 PBPK模型结合毒理基因组学的方法
Thiel等[35]将毒理基因组学与PBPK模型结合预测药物肝毒性和毒性阈剂量,提出了具有代表性的方法即基于PBPK模型的体外毒性数据体内转化(PBPK-based in vivo contextualization of in vitro toxicity data,PICD)法。PICD法利用基因表达谱数据库和功能富集分析方法,将人和大鼠的体外细胞实验所获的毒性数据,转化为定量化的体内毒性基因表达数据和关键生物学通路数据,并整合到人或大鼠的全身PBPK模型中,模拟药物产生毒性的基本分子机制,定量预测不同药物剂量或体内浓度下产生的药物体内毒性反应随时间变化的情况。他们使用体外细胞实验,发现硫唑嘌呤对细胞分裂周期25B蛋白、Wee1蛋白激酶和CHK1细胞周期检查点激酶(CHK1 checkpoint homolog),特别是对polo样激酶1(polo-like kinase 1)和细胞周期调控蛋白B1/B2均产生了显著的影响,然后使用基因芯片阵列测定不同药物浓度下肝细胞基因的变化,建立了硫唑嘌呤肝细胞浓度-基因变化模型;随后他们将该模型与人或大鼠的PBPK模型整合从而建立PICD方法,并构建硫唑嘌呤剂量-肝浓度-基因变化之间的关系。因此,可以依据硫唑嘌呤口服剂量的变化,得到硫唑嘌呤致肝毒性的基因和细胞变化,达到预测药物肝毒性的效果。此次研究预测结果,硫唑嘌呤在20.7 mg·kg-1剂量下会使细胞DNA复制和细胞循环受到抑制,进而引发药物肝毒性[36]。
这种研究思路可推广至其他药物的肝毒性预测中,有助于临床应用中药物不良事件的早期诊断;通过链接PBPK模型,PICD法还可以预测药物阈剂量,提供新的临床用药思路。通过选择合适的体外实验方法和建立PBPK模型,PICD法也可用于预测药物的肾毒性、心血管毒性和发育毒性等,应用范围广泛,发展空间大。
3.2 PBPK模型结合细胞分析的方法
在药物的安全性评估中,替代动物试验的方法正变得越来越重要,各种细胞模型已经被开发出来,如人类肝细胞系、新鲜分离的肝细胞、3D模型和器官芯片模型等细胞分析模型[37]。但细胞模型只考虑药物肝毒性的敏感性,可描述和预测肝毒性在体外系统的情况,尤其是考虑药物与细胞的蛋白质、脂质和基因的结合和分布,但忽视了体内暴露量和持续时间。而PBPK模型则可以模拟和预测药物在体内动态变化过程[38]。因此,研究者开始考虑将细胞模型和PBPK模型进行结合并预测肝毒性。
Paini等[38]构建了一种结合虚拟细胞分析(virtual cell based assay)和PBPK模型预测雌二醇肝毒性的方法。此方法使用HepaRG细胞作为人体肝细胞的替代品,细胞活性作为体外毒理学终点指标,并使用虚拟肝细胞作为肝毒性研究模型,虚拟肝细胞模型所需要的相关参数可从细胞实验和PBPK模型中获得,包括无影响浓度(no effect concentration)和杀伤率。他们将虚拟肝细胞的模拟结果和PBPK模型整合到KNIME数据工作平台,开发了一个用户友好的工具,用于预测肝毒性。这种方法强化了体内外药物暴露量与肝毒性之间的联系,可用于药物肝毒性剂量预测,以支持药物肝毒性的风险评估。
根据体外实验获得不同浓度下肝细胞毒性变化数据,建立药物浓度与肝毒性关系的数学模型,而PBPK模型可以模拟药物体内浓度动态变化过程,获得药物剂量和肝浓度的变化曲线[32],这2个模型融合计算,获得“药物剂量-体内肝浓度-肝毒性”之间的定量关系,即可对肝毒性进行预测。Yamazaki等[39]对肝毒性的预测方法就是采用了上述策略。他们在预测了53种不同化学物质的口服生物利用度(基于Caco-2细胞渗透性实验)后,利用日本Hazard Evaluation Support System(HESS)平台,拟合了口服生物利用度与未产生肝毒性药物阈浓度之间的定量关系。之后使用简化的大鼠PBPK模型估算药物的肝与血浆浓度比与药物口服剂量之间的关系,发现部分药物的PBPK模型预测的最大血浆浓度和血药浓度-时间曲线下面积(area under curve,AUC)与已报道的大鼠体内的无明显肝毒性的浓度一致,并且发现2-巯基苯并咪唑等几种化合物的最大血药浓度超过了引起肝毒性的阈值浓度,可能引发肝毒性反应。该方法证实了大鼠PBPK模型预测肝毒性的可能性,未来可推广至人体PBPK模型。
3.3 PBPK模型结合深度学习的方法
随着人工智能、深度学习方法研究的推进,相关研究方法也被应用到肝毒性的预测中,并取得不错的效果。如Alarecht等[40]提出了一种利用SVM分类器与PBPK模型预测肝毒性的新方法。他们选取了28种典型的肝毒性或非肝毒性化合物作为训练集,利用人类原代肝细胞和HepG2细胞对其进行体外细胞毒性实验,获得相关细胞毒性数据,包括不同孵育时间(24,48和72 h)的EC10(引起10%毒性效应的浓度)、EC50(引起50%毒性效应的浓度)和EC90(引起90%毒性效应的浓度);然后根据文献报道的训练集化合物的安全剂量和肝毒性剂量,利用PBPK模型预测得到相对应的体内暴露量(最大血药浓度Cmax、全血中Cmax和AUC等);然后将训练集化合物对应的体外毒性数据,体内暴露量以及该暴露量对应的临床结果(肝毒性/非肝毒性)输入SVM分类器,通过机器学习可以实现对肝毒性和非肝毒性训练集化合物的分离,而且通过计算药物在SVM向量空间的坐标,可以计算其肝毒性概率。因此,对于信息有限的化合物,该法可以直接根据化合物的体外细胞毒性数据,推测化合物肝毒性的特定概率所对应的血液浓度。最后,通过PBPK建模,血液浓度可以反推为相应的口服剂量,从而将口服剂量与一定的肝毒性风险联系起来,预测该化合物在特定概率下不产生肝毒性的可接受日摄取剂量。该研究也发现,将药物的全血Cmax以及48 h的EC10作为SVM分类器的输入参数时,该法能达到最佳的分离性能和准确度,其敏感性、特异性和准确性分别为100%,87.5%和93.3%,显示出较传统毒理学研究更优越的性能。未来该法可广泛应用于预测西药和中药成分的肝毒性,为药物的肝毒性研究提供新的思路。
3.4 PBPK模型结合DILIsym模型的方法
药物产生肝毒性的本质原因是引起肝细胞死亡[41],但肝细胞死亡与细胞内细胞器或蛋白质的变化有着密切的关系[42]。为提高预测准确性,Simulations Plus公司联合19家较大的制药公司、美国食品药品监督管理局(Food and Drug Administration,FDA)和学术界共同支持研发的一个DILI评估系统,PBPK模型结合DILIsym模型的方法(以下简称“DILIsym模型”)[43],这也是以PBPK模型为核心技术,基于药物对细胞器或某蛋白的干扰以预测药物肝毒性最为成功的应用。
DILIsym模型利用定量系统毒理学的原理,集PBPK模型、DILI机制数据与患者的个体差异为一体,预测待测药物的肝毒性和肝毒性生物标志物浓度随时间变化的情况[43-44]。目前,DILIsym模型整合了当今最为前沿的肝毒性机制数学模型,包括线粒体损伤、活性氧的产生(氧化应激)、胆汁酸瘀积、活性代谢物的产生、脂毒性和先天免疫反应的激活等,这些机制均会引起肝细胞凋亡或坏死,最终会引起传统生物标志物如GPT和非传统生物标志物如谷氨酸脱氢酶和miR-122等释放并进入体循环[45-46]。
DILIsym模型结合模拟体内药物动态浓度变化的PBPK模型,根据体外获取的肝毒性机制的特征参数,使用所配套的SimPops®软件构建虚拟人群,用来预测人群中的肝毒性反应,并得到肝毒性反应的个体差异,表现为肝损伤生物标志物的体内动态变化[47-48]。DILIsym型的概念如图1,可以呈现多维度肝生理变化学,包括药物和细胞相互作用、药物对肝整体的影响作用、药物的全身分布和代谢的变化,以及药物引起肝毒性反应后人体生物标志物的变化[49]。
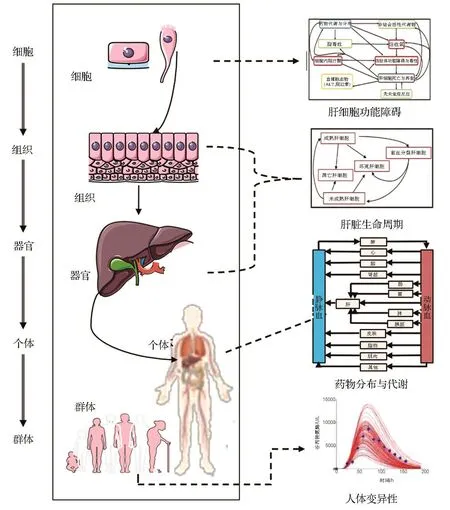
图1 DILIsym模型概念[49].左侧内容代表药物在不同生理维度的变化(即细胞、组织、器官到人体层面);右侧分别代表相应维度下药物对肝细胞、肝和人体产生的影响。体外细胞实验体现药物对肝细胞功能的影响,PBPK模型体现药物在体内的浓度变化,SimPop®软件体现人体肝功能生物标志物的变异性。DILIsym模型可以达到直接预测药物肝毒性的功能(即肝损伤生物标志物的浓度变化).
目前,DILIsym模型主要应用于回顾性研究,以验证该模型的预测准确度。为验证DILIsym模型预测肝毒性的前瞻性能力,目前有几项临床研究正在开展中。DILIsym模型对药物TAK-875是否产生DILI进行了回顾性预测研究。TAK-875的体外细胞毒性机制数据(抑制胆汁酸转运蛋白和线粒体电子传递链酶)结合PBPK模型和245个虚拟2型糖尿病患者,经过DILIsym模型预测,发现不同药物剂量下PBPK模拟结果与实际结果拟合较好(血浆中AUC和Cmax的观察值与模拟值比值<1.5)。而且也发现虚拟患者的GPT浓度具有差异,尤其是200 mg·d-1的剂量下,虚拟患者的GPT浓度>上限3倍的概率为16/245(6.5%),与实际临床试验的结果(4.0%)接近,说明DILIsym模型可以预测药物是否导致人肝毒性,而且也可模拟临床试验的结果[50]。
而且,DILIsym模型也可有效避免预测结果出现种属差异。曲格列酮的临床研究表明其具有肝毒性,但是在动物实验中却未被发现。DILIsym模型根据体外肝细胞毒性数据定量预测曲格列酮肝毒性时,发现临床剂量在20~60 mg·d-1时,虚拟受试者出现GPT超过正常值上限3倍以上的比例是0.3%~5.1%,接近于临床上观察到的比例(1.9%)。同时预测结果也发现,胆汁酸在人体内的升高幅度远高于大鼠,这与大鼠的胆汁酸具有亲水性强和毒性小有关,所以动物实验结果才会与临床试验结果相反[9]。从曲格列酮也可看出,虽然动物实验还是进行非临床肝毒性研究的重要方法,但种属差异会影响最终结果。而DILIsym模型的预测结果不仅可有效避免种属差异问题,有效提高预测的准确性,而且可为解决待测药物待解决的肝毒性机制问题提供参考,具有一举多得的优点。
在药物肝毒性的预测中,许多同类药物由于结构类似,常被认为具有类似的肝毒性,但事实并非如此,如托卡朋和恩他卡朋。DILIsym模型预测结果发现,托卡朋使用推荐剂量时,2.2%虚拟受试者会出现GPT显著升高,与临床观察到1.3%~5.0%的频率相符;而恩他卡朋未发现引起GPT显著升高。且DILIsym模型也发现恩他卡朋的肝清除速率快于托卡朋,托卡朋的线粒体解偶联作用强于恩他卡朋,且托卡朋的肝浓度比恩他卡朋高出3倍,因此才会出现两者结构类似但肝毒性相差较大的结果[51]。
另一方面,DILIsym模型由于能够很好地描述肝细胞的生理过程,如肝的再生过程和细胞凋亡或坏死后生物标志物的释放等,因而也能解释临床上发现的肝损伤生物标志物变化时肝损伤的具体情况。恩托利莫(entolimod)作为一种抗肿瘤药物,在健康志愿者的早期安全性试验中发现,该药可引起受试者的GPT和GOT显著升高,甚至>1000 U·L-1,所以该药的研发一度中止。但通过DILIsym模拟预测发现,该药造成的肝细胞坏死数量≤整个肝细胞数量的5%,<肝移植时捐赠者的肝切除比例,不会带来很大的健康风险。因此,该药的开发得以继续进行[52]。这也证实了DILIsym模型可以有效的避免药物研发中的“滥杀无辜”现象的出现。
DILIsym模型所使用的数据是细胞器水平的毒性数据,依据药物对细胞内部结构的影响,提前预知药物致肝损伤的前期变化,达到早期预测的效果。同时,DILIsym模型也可为未知药物致DILI的机制提供参考。而且DILIsym模型可以预测肝细胞再生过程、生物标志物释放和细胞凋亡或坏死程度,因此能有效预测一过性肝损伤,避免出现过度预测。随着模型的不断优化,未来DILIsym模型也许可以替代部分临床试验,减少药物研发对临床试验的依赖,简化药物研发流程,加快药物上市流程。但DILIsym模型也有缺陷,如缺乏特殊人群模型,如儿童和老年人模型;缺乏其他肝损伤机制的参数。
基于PBPK模型的预测方法可以有效预测药物是否产生DILI和毒性口服阈剂量,而且在预测化合物的肝毒性方面具有很大优势:不依赖化合物结构,可以避免出现结构相似的化合物预测结果也相似的问题;有效避免预测结果出现种属差异的情况,提高药物研发效率;也可以确定药物的可接受日摄取剂量,有效提高药物临床试验的成功率和使用安全性。但是由于细胞实验和建模过程的复杂性,这类方法失去了QSAR模型简单易行的优点,不适用于快速筛选药物。
4 结语
DILI是一种广受关注的严重药物不良反应,其预测方法的研究可以更早获知药物的肝毒性,代替动物实验,提高临床试验和临床用药安全性。因此,数学模型预测DILI方法的快速发展为全球药物研发和临床使用提供巨大的帮助。目前正在研究中的3类数学模型DILI预测方法:基于QSAR模型的方法、基于毒理基因组学模型的方法和基于PBPK模型的方法,从不同的角度提出了可替代传统体内实验方法,促进药物研发和使用的进步。因此,药物研究者可以在不同阶段根据需求选择合适的方法。
目前,DILI预测方法的发展仍面临许多挑战。QSAR方法和其他需要使用计算机算法的方法在使用中既需依靠所选化合物的性质和数量,也需依靠算法的普适性和准确性,所以无法判别哪种化合物和算法更适合DILI预测,预测结果的准确性也无法保证;毒理基因组学所依赖的基因组学测序技术面临升级困境,且缺乏数据信息挖掘的新技术;而PBPK模型的建立依靠大量体内外数据,如果药物体内变化过程无法明确,或缺乏特殊人群的体内数据,则无法建立合适的PBPK模型,从而降低预测结果准确性。未来数学模型预测DILI方法的发展有赖于肝毒性机制和基础计算机算法研究的进步,也需要依靠技术和实验设备的发展。
猜你喜欢
昆明医科大学学报(2022年2期)2022-03-29 00:52:18
中学生数理化·中考版(2021年12期)2021-12-31 03:24:40
中学生数理化·中考版(2021年11期)2021-12-06 07:29:16
小哥白尼(野生动物)(2019年5期)2019-08-27 00:53:38
中学化学(2017年6期)2017-10-16 17:22:41
癌变·畸变·突变(2016年3期)2016-02-27 06:15:36
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:10
哈尔滨医药(2015年4期)2015-12-01 03:57:54
发明与创新(2015年33期)2015-02-27 10:40:02
郑州大学学报(理学版)(2014年2期)2014-03-01 04:20:55
