
结直肠癌发生过程中普拉梭菌丰度变化的研究*
2021-07-13 12:06钟华戈严林海吴昌淘唐卫中
广西医科大学学报 2021年6期
钟华戈,严林海,陈 伊,吴昌淘,唐卫中
(广西医科大学附属肿瘤医院胃肠外科,南宁 530021)
结直肠癌是男性第三大、女性第二大常见癌症,且致死率排在所有癌症的第三位,近年来结直肠癌在我国的发病率也呈明显上升趋势[1]。研究表明,早期结直肠癌患者经过治疗可以获得满意生存,但是大部分患者就诊时病变已经处于进展期甚至晚期,错过了最佳治疗时间,再者因原发肿瘤无法切除,化疗药物耐药和肿瘤复发等因素,从而导致晚期结直肠癌预后不佳[2-4]。结合目前早期筛查可有效遏制结直肠癌高发,因此,寻找一种早期发现结直肠癌的方法显得十分有必要。
结直肠癌的形成是个复杂且多样的过程,成因包括体细胞分子突变、饮食、环境、微生物和宿主免疫等。随着对人类微生物研究的越来越深入,人类微生物组参与机体代谢,疾病的发展已被证实[5]。在人体内,结直肠中的肠道菌群是维持肠道微生态系统稳定的重要组成成分,主要通过直接或间接参与了免疫调节、物质代谢和消化吸收等过程,从而保护肠黏膜、维持肠道稳态和机体正常功能以及抵抗疾病等[6]。Zouggar 等[7]研究表明,肠道菌群随着宿主的生活和饮食习惯进行动态调节,与结直肠癌的发生密切相关,当其结构和功能受到破坏时通过破坏肠黏膜上皮细胞、参与宿主机体代谢和产生致癌物质等方式诱发结直肠息肉和癌变。此外,研究表明,肠道微生物群在“腺瘤-癌序列”的形成中起到了重要的作用[8]。结直肠腺瘤发展为肠癌往往需要较长时间,这为在筛查过程中发现癌前病变并予以治疗提供了时间窗,从而防止其癌变并使患者获得长期生存。因此,肠道道微生物群在结直肠癌的起始可能提供对肿瘤发生和早期预防潜在新方向。本研究通过微生物信息数据库结合结直肠腺瘤患者宏基因组测序,观察和验证特征菌群变化,为通过肠道菌群作为生物标志物筛查和治疗干预直肠癌提供实验依据。
1 材料与方法
1.1 样本采集 选取广西医科大学附属肿瘤医院收治的35 例结直肠腺瘤患者及35 例健康对照,均行肠镜检查,年龄40~69 岁,在广西本地居住5 年以上,两个月未服用抗生素、酸奶,并排除其他肠道疾病。于患者镜检2 个月后,取患者粪便样本中段于无菌冰盒,分装于2 mL EP管中,放置于-80 ℃冰箱中保存。结直肠腺瘤患者与健康对照一般资料比较,差异无统计学意义(P>0.05),具有可比性。本研究已取得本院医学伦理委员会批准,所有研究对象均签署知情同意书。
1.2 粪便样本肠道菌群DNA提取 取200 mg粪便样本,加入2 mL EX 缓冲液。取1.2 mL 粪便裂解物,70 ℃加热5 min,1 500 r/min离心1 min,取200 μL上清液于含有1.5 mL 蛋白酶K 的微量离心管中;70 ℃下孵育10 min,加入200μL 75%乙醇,混匀;600 μL裂解物通过QIAamp离心柱,离心;向离心柱中加入500 μL Buffer AW2,1 500 r/min 离心1 min洗脱DNA。
1.3 粪便DNA宏基因组测序及文库构建 严格按照TruSeq Nano DNA Sample prep Kit(Illumina)说明书进行全基因组测序和文库构建。NanoDrop 测定gDNA 浓度,琼脂糖凝胶电泳检查gDNA 的完整性,样本质检后,使用Covaris S220 Sonicator 将gDNA 打断至300~400 bp,纯化,产物末端经过修补成平末端,用磁珠进行片段筛选;在3’末端加A。此片段的两端分别与通用的接头相连接,其中一个接头带有特异性的barcode序列,用以区分样本的来源。连接产物经纯化去除连接不完整的产物以及接头自连产物,用与接头序列互补的通用引物进行PCR 扩增,产物纯化,琼脂糖凝胶电泳质检,Qubit测定浓度,2100 Bioanalyzer测定片段长度,构建gDNA 文库,用Agilent 2100 Bioanalyzer 进行文库质检,并对腺瘤组及健康对照进行菌群丰度的比较分析。
1.4 健康对照—腺瘤患者—腺癌患者菌群差异分析 人类肠道宏基因组数据库GMrepo是常用于肠道微生物菌群研究的数据库之一,是由研究人员阅读海量文献以特定的图形语言描述众多微生物菌群改变和相互关系。GMrepo微生物数据库的信息包括253个微生物测序项目的58 903个人体肠道样本,与92种人类表型相关。本研究选取数据库中健康对照、腺瘤患者、腺癌患者各30例,通过Fisher精确检验方法对比3组微生物富集情况。
1.5 统计学方法 本项目分别采用MetaPhlAn25与HUMAnN26 生信分析软件进行分析。MetaPhlAn2与HUMAnN2是由美国国立卫生研究院(NIH)发起的人类微生物组计划(HMP)发布的宏基因组学官方分析软件对菌群丰度的分析与功能注释进行分析。后期数据处理与统计分析则采用LEfSe7、STAMP8、QIIME9等国际公认的宏基因组学分析软件与R 包(heatmap,ggplot2)进行分析。采用Wilcoxon 秩和检验、Spearman 相关性、Fisher 精确检验方法等方法来评价肠道菌群相关的差异分析。以P<0.05为差异具有统计学意义。
2 结果
2.1 GMrepo分析健康对照—腺瘤患者—腺癌患者菌群丰度改变 对健康对照、肠道腺瘤患者、结直肠癌患者的粪便菌群原始丰度进行GMrepo分析发现,健康对照和肠道腺瘤患者之间有4 个菌属丰度存在显著差异。肠道腺瘤患者和结直肠癌患者之间有6个菌属丰度存在显著差异。健康对照和结直肠癌患者之间有8个菌属丰度存在显著差异。健康对照、肠道腺瘤患者、结直肠癌患者3 组间98 个菌属中有3个菌属存在显著丰度差异(P<0.05),其中包括瘤胃球菌科(包括瘤胃球菌科和粪杆菌属)、双歧杆菌科、梭菌科,见图1。
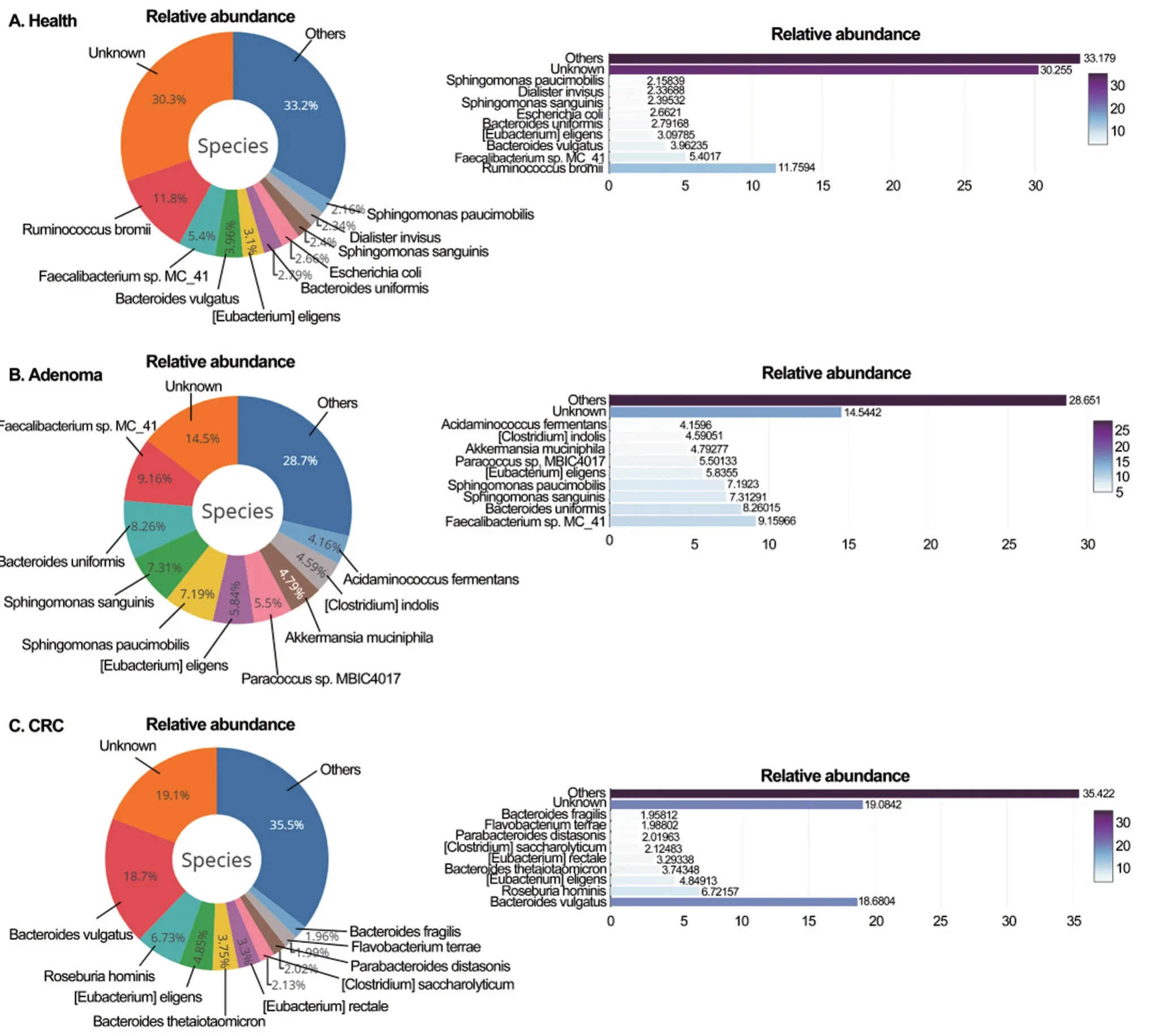
图1 健康对照、肠道腺瘤患者和结直肠癌患者微生物菌群丰度在菌种水平上的差异
进一步对腺瘤和结直肠癌患者进行具体菌群差异分析,发生腺瘤到结直肠癌过程中患者普拉梭菌和双歧杆菌的相对丰度较低,而牙周梭杆菌的相对丰度较高。采用随机森林法对两组进行最优区分的重要物种进行识别,其中最重要的菌种为普拉梭菌,其次是牙周梭杆菌和双歧杆菌,见图2。
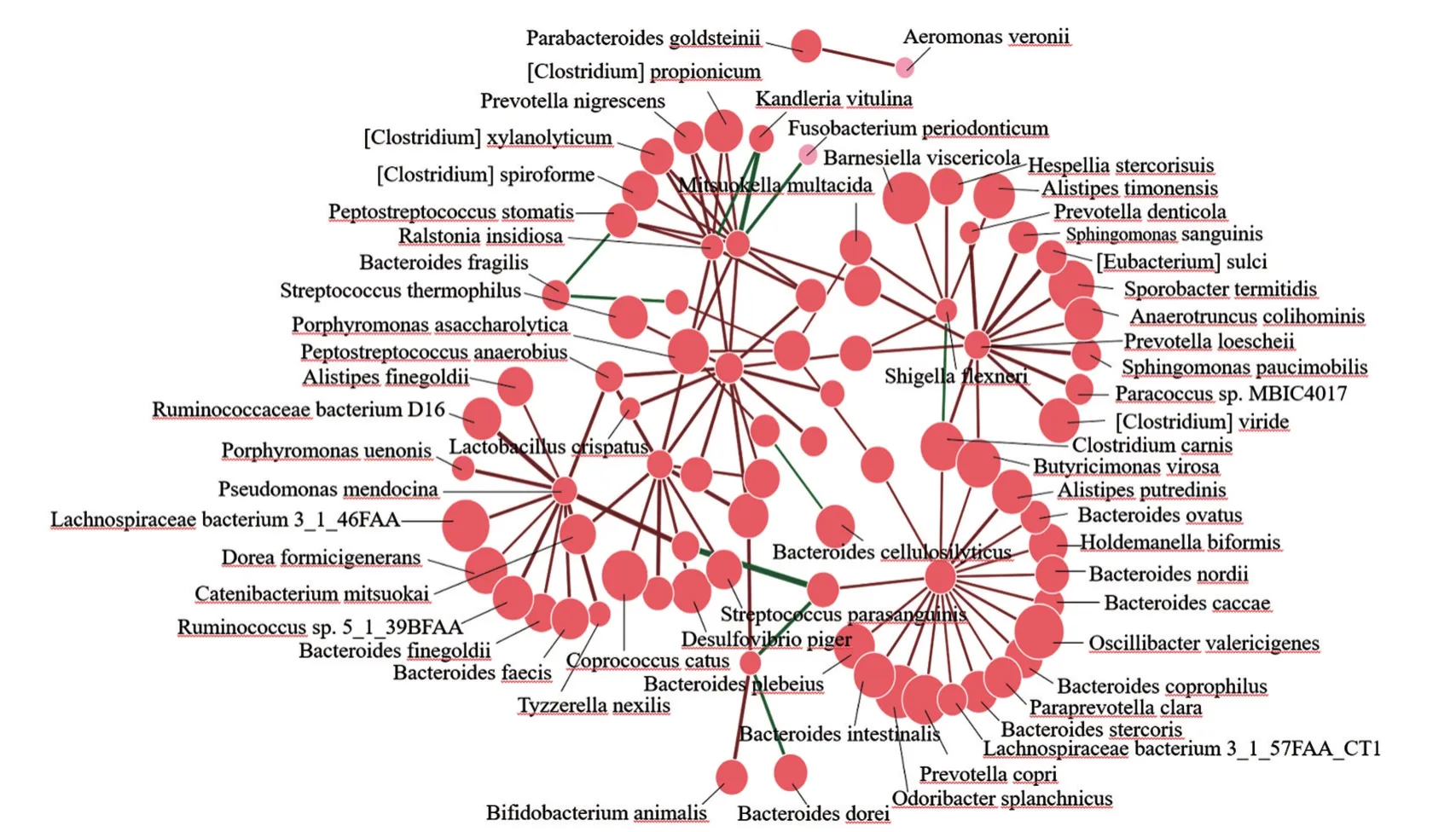
图2 腺瘤、结直肠癌患者菌群GMrepo差异分析
2.2 肠道微生物宏基因组学测序验证腺瘤患者与健康对照菌群差异 腺瘤患者的菌群丰度与健康对照比较,差异有统计学意义(P<0.05),见图3。MetaPhlAn、HUMAnN 分析发现,腺瘤患者与健康对照在细菌微生物菌门水平上也存在显著的差异,对每个重要分类单元的LEfSe 分析后进行LDA 评分。采用随机森林法对两组进行最优区分的重要物种进行识别,发现两组肠道菌群在门水平上,放线菌门有明显差异(P=0.002)。
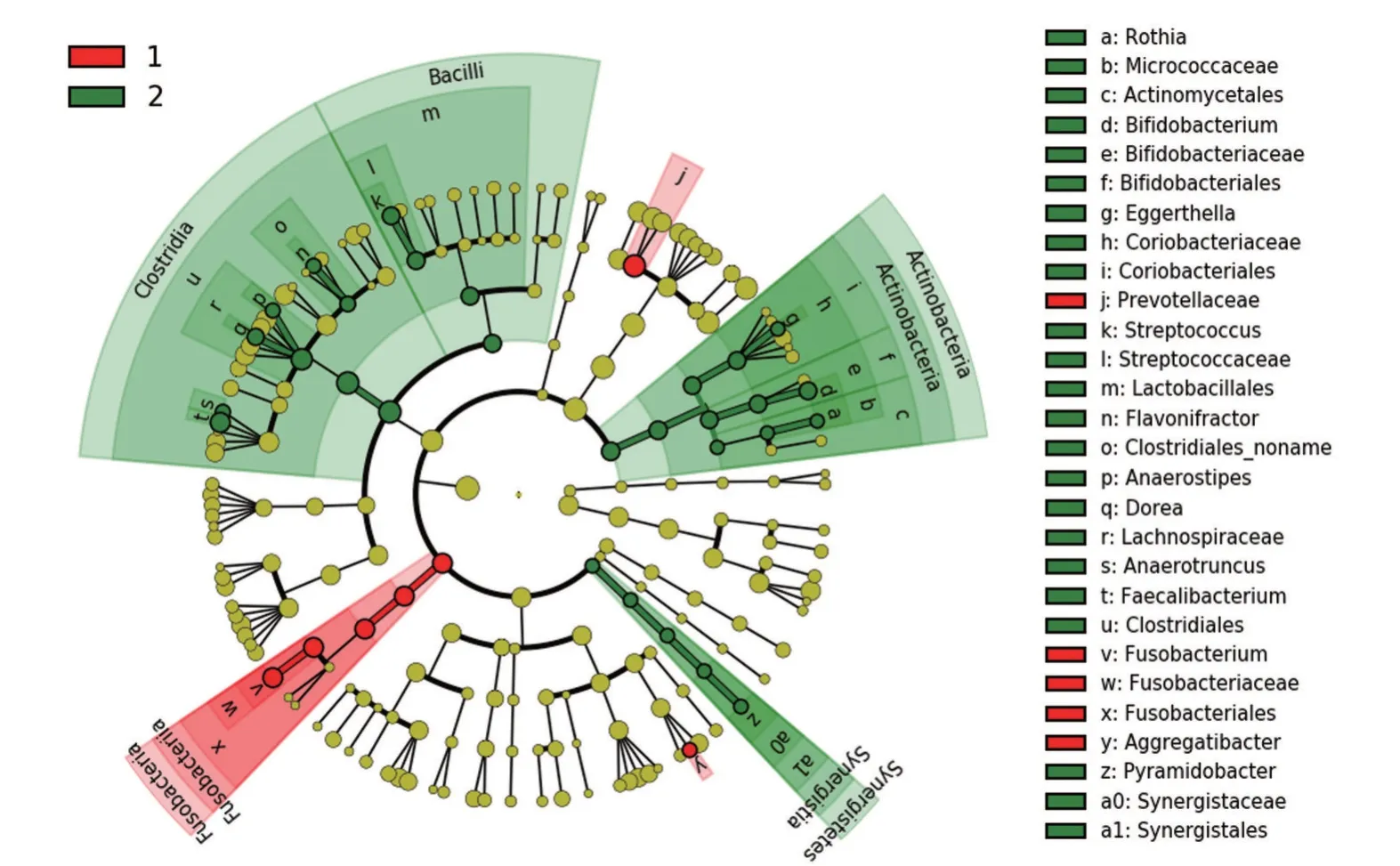
图3 腺瘤患者与健康对照菌群丰度比较
2.3 腺瘤患者与健康对照肠道微生物在菌属水平上的差异 健康对照和腺瘤患者肠道菌群中普拉梭菌、罗氏弧菌、双歧杆菌、唾液链球菌有显著差异(均P<0.05),见图4。
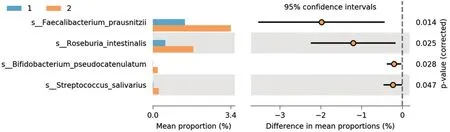
图4 4种水平腺瘤患者与健康对照菌群丰度比较
3 讨论
结直肠癌的发生因素包括体细胞基因突变,生活环境,饮食结构,宿主免疫和肠道微生物组成等,只有25%的患者有显著的家族遗传倾向,6%患者的癌症发生与致癌基因突变存在显著的关系[9]。生活习惯对结直肠癌的发生有着重要的意义。但是,以饮食习惯为例,并不是每个有高脂、多肉和低纤维饮食习惯的人必然会发展为肠癌。有证据指出结直肠癌的产生是由于受饮食等因素的影响整体肠道菌群的组成逐渐变化与基因的突变导致抑癌基因沉默共同导致的[10]。
口腔、呼吸道、肠道、泌尿生殖道等都存在着不同种类和数量的稳定共存微生物群,称为定居菌群。在定居菌群中,数量最多及种类最丰富的是肠道微生物,人类结肠中有非常多样化和复杂微生物菌群区包括估计有100万亿的细菌,与病毒、细菌和真菌一起超过1 000 种异构物种(约400 万个基因)。肠道微生物的细菌基因组集是人类基因组基因的150倍,随着近几年人类微生物研究的热潮,人类微生物组参与机体代谢,疾病的发展已被证实,实验研究表明微生物发酵产物(如丁酸盐)和微生物活化植物化学物质(例如聚—酚类)具有广泛的抗肿瘤效果,能抵抗肿瘤发生信号通路如组蛋白脱乙酰酶抑制促进细胞凋亡,抑制增殖并阻滞肿瘤转化肠道微生物可以通过许多方面影响结直肠癌的发生发展[11-14]。
通过GMrepo 网络分析和宏基因组学验证,本研究发现,健康—腺瘤—腺癌3 组的肠道菌群丰度存在明显差异。属水平上健康对照组有3 种细菌(双歧杆菌、链球菌、粪杆菌)明显高于实验组,而双歧杆菌和粪杆菌均为益生菌,其中属于放线菌门的双歧杆菌参与了免疫系统的成熟,是已知最具代表性的人肠道益生菌[15]。
此外,本研究发现普拉梭菌丰度降低,是发生在健康—腺瘤—腺癌全程中的事件,普拉梭菌属于厚壁菌门梭菌科。约占肠道粪便细菌总数的5%,是柔嫩梭菌亚群的主要成员之一,普拉梭菌不仅在健康对照和肠道疾病患者肠道及粪便中分布存在明显差异(P<0.05),并在肠道疾病中起到了肠道黏膜屏障保护及炎症抑制等重要作用。它也是结肠中最重要的产丁酸菌,这种共生细菌已被认为是人类健康的生物指标,一旦它的种群减少,会使得腺瘤患者有更大的可能性发展为腺癌[16]。普拉梭菌与8 种肠道炎性代谢产物有相关性,提示这种细菌在参与宿主多种代谢过程中有着很重要的作用,其改变导致的微生态失衡与机体炎症相关,虽然目前暂无普拉梭菌制成微生态制剂而直接应用于临床,但有研究表明普拉梭菌及其上清具有抗炎效应,可明显改善肠道炎症[17]。
因此可以推断肠道菌群丰度显著改变可能与肠道炎症反应和免疫紊乱相关,而较高的炎症反应与免疫紊乱使腺瘤患者相对健康对照来说有更大的可能发展为结直肠癌,其中普拉梭菌丰度降低,有可能是加速这一过程的重要影响因素。
猜你喜欢
质量安全与检验检测(2022年2期)2022-11-13
临床儿科杂志(2022年9期)2022-09-01
中国畜牧杂志(2022年7期)2022-07-13
当代水产(2022年1期)2022-04-26
当代水产(2022年2期)2022-04-26
中国饲料(2022年5期)2022-04-26
医学食疗与健康(2022年2期)2022-04-23
环球时报(2017-03-15)2017-03-15
作文周刊·小学一年级版(2016年7期)2016-08-11
数学大世界·小学低年级辅导版(2010年9期)2010-09-08

- 广西医科大学学报的其它文章
- Musashi2 gene expression in lung squamous cell carcinoma and its effect on the maintenance of CD44v6(+)lung cancer stem cells mediated by Notch1 signaling pathway
- 持续隐匿性乙肝病毒感染树鼩肠道菌群变化研究*
- TREM1/TREM2在小鼠呼吸机相关性肺损伤中的表达*
- 色素上皮衍生因子联合5-氟尿嘧啶抑制结肠癌细胞凋亡和血管生成的作用研究*
- 葡醛内酯对异烟肼和利福平所致肝损伤小鼠肝组织自噬水平的影响*
- 基于网络药理学和分子对接探讨鸡血藤治疗冠脉微循环疾病机制*