
三叶木通雌雄花芽转录组测序及分析
2021-07-12 04:23:44陈发菊
三峡大学学报(自然科学版) 2021年4期
田 天 项 明 陈发菊
(三峡大学 生物技术研究中心,湖北 宜昌 443002)
三叶木通(Akebiatrifoliate)是木通科(Lardizabalaceae)木通属(Akebia Decne.)落叶木质藤本植物,民间又称八月炸、八月瓜.三叶木通花序为总状花序,下部生有1~2朵雌花,上部开有15~30朵雄花.三叶木通早期均为两性花结构,在发育的后期雌蕊和雄蕊会选择性退化而形成单性花[1].
高通量测序技术是近些年来发展出的新技术,广泛用于SSR标记的开发鉴定[2],各种生理活动的代谢途径以及基因分析[3],也被广泛用于性别相关基因的挖掘,韦丽君等[4]发现,生长素、WRKY和MYB转录因子相关的基因与木薯性别决定相关.研究表明簸箕柳的转录测序分析发现33条差异表达基因定位到十九号染色体上[5],王萌通过转录组测序发现AG与FT基因可能对核桃的开花调控具有重要作用[6].何晓琳通过对蓖麻的不同种系的转录组对比分析确定了杂种优势的基因及代谢通路[7].闫冬春在南瓜转录组找到了一个影响雌雄花发育的关键基因CmACO1[8].在山核桃的雌雄转录组分析中,研究者聚焦到了MADS-box基因家族,推测AP1和SEP亚家族基因可能与雌雄花的形成有紧密联系.这些研究充分说明利用转录组测序来获得植物性别分化的相关基因是可行的.
本实验通过三叶木通的雌雄花芽进行转录组测序,利用现有的数据库与测序结果进行比对,并对雌雄花芽的差异基因进行验证分析,为探究三叶木通性别分化机制奠定基础及利用基因工程培育新品种提供方向.
1 材料与方法
1.1 实验材料
测序材料三叶木通雌雄花芽采自湖北省神农架自然保护区.以三叶木通雌花芽为对照组,记为MT1,以三叶木通雄花芽为实验组,记为MT2.实验材料采集之后立刻过氮处理,并保存于-80℃冰箱,转录组测序样本重复3次,分别为MT1-1,MT1-2,MT1-3,MT2-1,MT2-2和MT2-3.
1.2 三叶木通总RNA提取、文库制备及测序
采用Trizol试剂盒(Invirtrogen,CA,USA)提取三叶木通雄花芽和雌花芽总RNA,紫外分光光度计和Nanodrop软件检测RNA浓度与纯度,1%琼脂糖凝胶电泳检测总RNA质量和完整性.
按照Illumina公司的文库制备实验流程制备文库,样本总RNA使用连接有Oligo(dT)的磁珠富集真核生物mRNA,然后被随机打断成短片段,用随机引物法合成cDNA第一链,再合成cDNA第二链,最后纯化回收双链产物,利用T4 DNA连接酶和KlenowDNA聚合酶进行进一步处理,最后用USER酶降解cDNA第二链进行PCR扩增获得测序文库.文库质检合格后采用Illumina NovaseqTM6000 进行测序,测序读长为双端2*150 bp(PE150).
1.3 质量控制、组装统计、功能注释与表达分析
首先,使用Cutadapt[9]和perl脚本去除低质量碱基和未确定碱基,然后使用FastQC(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/)通过有效数据的Q20,Q30和GC含量验证序列质量,后续分析均基于高质量的有效数据.使用Trinity 2.4.0[10]进行转录组序列的组装,将转录本中的序列称为transcript.选择其中最长的转录本作为Unigene.组装好的Unigenes通过DIAMOND[11]软件与各个数据库进行对比和功能注释.
1.4 三叶木通雌雄花芽差异基因的筛选
根据Patro[12]的方法计算TPM[13]衡量所有Unigenes表达水平,利用package edgeR[14]对Unigenes进行差异表达分析,然后通过log2>1(log2 fold change值,差异倍数)或者log2<-1并且具有统计学意义(P<0.05)进行筛选,获得三叶木通雌雄花芽中的差异基因.
1.5 三叶木通雌雄花芽差异表达基因的qRT-PCR验证
使用Trizol reagent试剂盒提取三叶木通总RNA,采用逆转录法(Takara)获得cDNA.依据转录组测序结果,随机筛选10条差异表达明显的基因,进一步分析筛选得到细胞分裂素以及丙酮酸相关的基因9个.通过实时荧光定量PCR仪(CFX96,BIO-RAD)以及SYBR qPCR试剂盒(南京诺唯赞生物科技有限公司)进行qRT-PCR实验.通过NCBI BLAST设计qPCR引物,由华大基因股份有限公司合成.qRT-PCR引物见表1,产物长度100~250 bp.选择RG5(EF-α)作为内参基因[15],上游引物为GTCTCCCTCTTCAGGATGTTTAC,下游引物为GACAACCATACCAGGCTTGA.其中前10对引物用来验证转录组数据的可靠性,后9对引物为代谢通路引物.

表1 实时荧光PCR引物
2 结果与分析
2.1 三叶木通转录组数据测序及序列组装
采用Illumina测序平台技术对三叶木通样本进行转录组测序,共得到49.84 Gb的原始数据(raw data).过滤掉不合格的序列,共得到49.06 Gb有效数据,占原始数据的98.43%,组装后GC含量与长度分布如图1~图2所示,每组数据质量见表2.对过滤后的数据进行Trinity组装,去除冗余后得到17 439个Unigene,通过六大数据库注释,所有基因均被一个或者更多的数据库注释到(见表3).

图1 Unigene组装长度统计 图2 Unigene的GC含量统计

表2 过滤前后的Reads统计

表3 Unigene功能注释统计
去除测序接头和过滤掉不合格序列后得到有效数据(clean data),去除测序reads接头序列、截短后长度小于100 bp的序列、N含量大于5%的序列后统计GC含量.采取所有样本混合组装的策略,将所有样本进行归一化得到Unigene(即各个样本所有基因进行去冗余去重复后得到的唯一序列),然后使用长度、GC含量以及N50作为标准对Unigene组装质量进行评判.N50是将所有组装结果按照长度从高到低排列后,达到总长度一半时的长度.选取权威的NCBI_nr、GO、KEGG、Pfam、Swiss-Prot和eggNOG 6种数据库,使用软件DIAMOND添加相应的功能注释,再通过RSEM软件计算各基因的表达水平,然后通过R语言对差异基因进行表达分析.
2.2 三叶木通Unigene的GO和KEGG功能注释
2.2.1 三叶木通Unigenes的GO功能分析
对三叶木通的Unigenes进行GO功能富集.共注释到75 631种不同的GO功能,其中生物学过程(Biological Process)占比最多,涉及到29 332个不同的功能,而细胞组分(Cellular Component)共有个26090个功能被注释到,富集到分子功能(molecular_function)共有23 929个,同一个Unigene可以对应多种不同的功能,按照功能注释富集的Unigenes如图3所示.
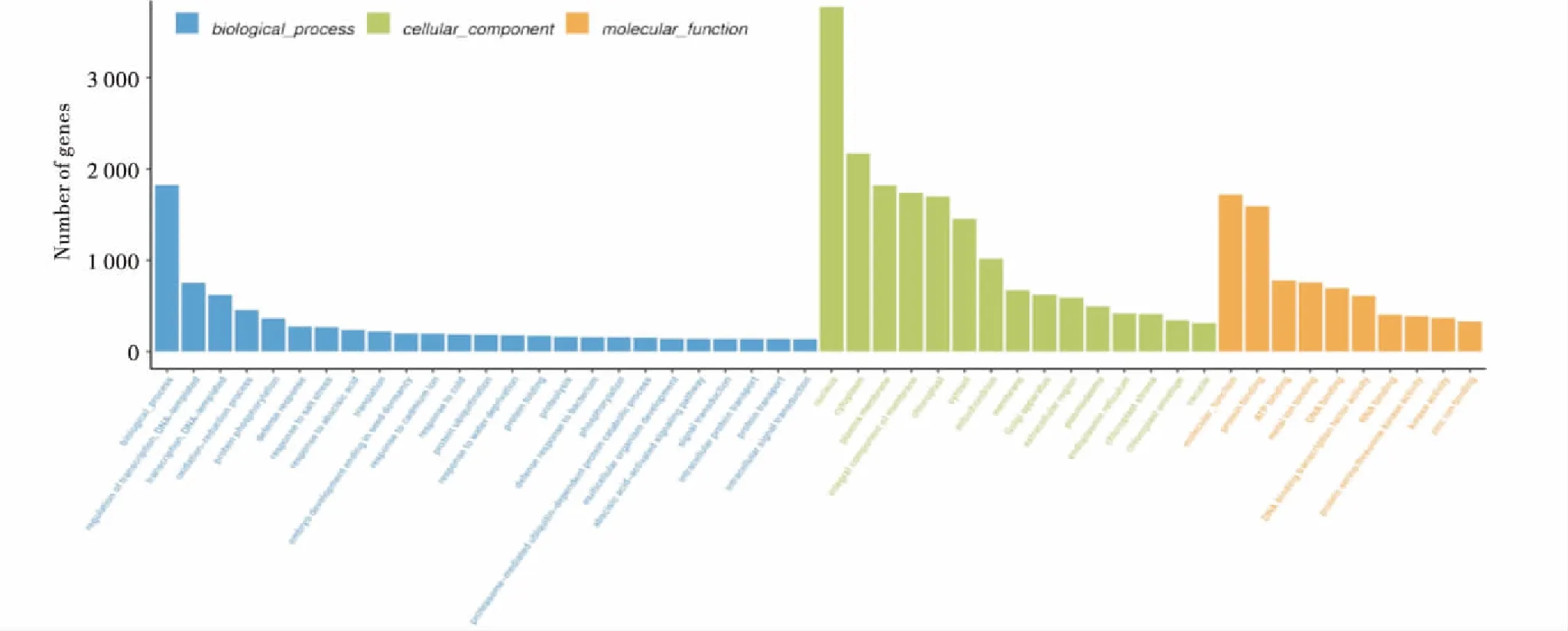
注:1~25:生物过程;26~40:细胞组分;41~50:分子功能.1.生物过程;2.DNA转录调控;3.DNA转录;4.氧化还原过程;5.蛋白质磷酸化;6.防御反应;7.对盐胁迫的反应;8.对脱落酸的反应;9.转化;10.胚发育结束于种子休眠;11.对镉离子的反应;12.对冷胁迫的反应;13.蛋白质泛素化;14.对水分缺乏的反应;15.蛋白质折叠;16.蛋白质水解;17.对细菌的防御反应;18.磷酸化;19.蛋白酶体介导的泛素依赖性蛋白质分解代谢过程;20.多细胞生物发育;21.脱落酸激活的信号通路;22.信号转导;23.细胞内蛋白质转运;24.蛋白质转运;25.细胞内信号转导;26.细胞核;27.细胞质;28.质膜;29.膜的组成成分;30.叶绿体;31.细胞质;32.线粒体;33.膜;34.高尔基体;35.胞外区;36.胞间连丝;37.内质网;38.叶绿体基质;39.叶绿体包膜;40.液泡;41.分子功能;42.蛋白质结合;43.ATP结合;44.金属离子结合;45.DNA结合;46.DNA结合转录因子活性;47.RNA结合;48.蛋白质丝氨酸/苏氨酸激酶活性;49.激酶活性;50.锌离子结合.
2.2.2 三叶木通的KEGG功能分析
对三叶木通的雌雄花芽进行KEGG pathway聚类分析,共注释到10 017个Unigenes,聚焦到20个生物途径上.其中显著富集的通路(如图4所示)主要是代谢途径和遗传信息处理,具体为碳水化合物,脂质,氨基酸的代谢,蛋白质折叠、翻译、分类与降解,淀粉和蔗糖代谢等.在对通路基因富集分析后发现,许多基因与生长素、细胞分裂素相关的通路密切联系.
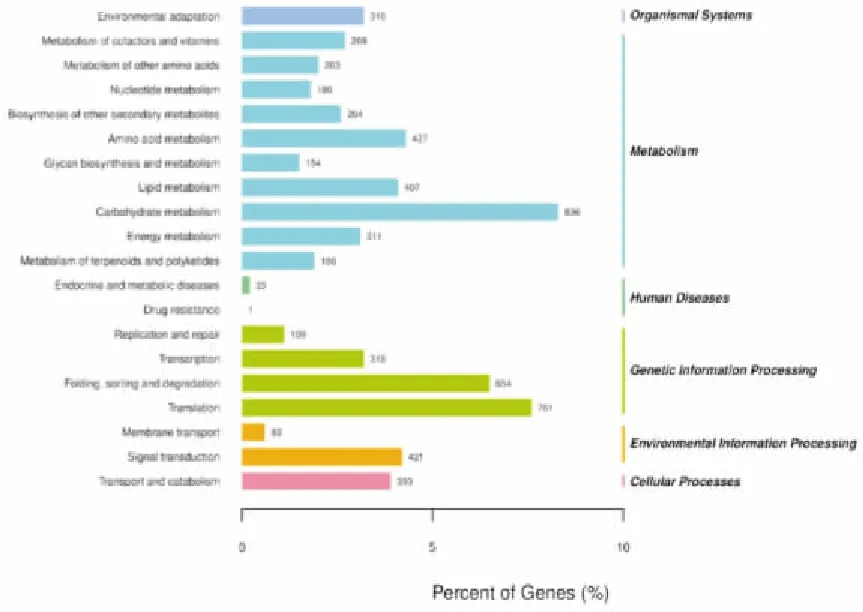
注:1:有机体系;2~11:代谢;12~13:人类疾病;14~17:遗传信息处理;18~19:环境信息处理;20:细胞过程.1.环境适应性;2.辅助因子和维生素的代谢;3.其他氨基酸的代谢;4.核苷酸的代谢;5.其他次生代谢产物的生物合成;6.氨基酸的代谢;7.糖类的生物合成和代谢;8.脂质的代谢;9.碳水化合物的代谢;10.能量代谢;11.萜类和聚酮化合物的代谢;12.内分泌和代谢疾病;13.抗药性;14.复制和修复;15.转录;16.折叠、分类和降解;17.翻译;18.膜转运;19.信号传导;20.转运和分解代谢.
2.2.3 三叶木通雌雄花芽差异基因富集分析
对转录本进行筛选分析后,以雌花为对照组,发现雄花中一共有2 579个Unigenes表达上调,2 793个Unigenes表达下调.然后利用R语言进一步对这些差异基因绘制火山图(如图5所示).通过差异基因聚类分析,可以判断基因在不同实验条件下调控模式的聚类模式,根据样品基因表达谱的相近程度绘制了差异基因聚类热图(如图6所示),为了更好地直观反映聚类表达模式,将差异基因TPM通过Z值方式进行基因表达展示.其中横坐标为样本,纵坐标为基因,不同的颜色表示不同的基因表达水平.

图5 三叶木通差异基因火山图

图6 三叶木通差异基因热图
2.2.4 三叶木通雌雄花芽差异基因的q-PCR验证
通过对差异基因进行筛选,选取了10个基因进行 了qRT-PCR验证,这些基因均在雄花芽中产生了高表达,与测序结果一致,说明转录组数据真实可靠.同时,对这些差异基因进一步挖掘分析表明,与花发育相关的MADS-box家族的基因与Unigenes的部分结果具有较高的重合度,从NCBI上已公布的三叶木通AG、SEP3、AP3序列进行了克隆并同时进行了序列比对,结果表明碱基重合度很高(如图7所示).

注:**代表差异性极显著,P<0.01
2.2.5 三叶木通的差异基因富集性分析
将三叶木通雌花与雄花的差异表达基因与GO以及KEGG数据库比对分析,结果与Unigenes差异主要在细胞组成部分.而差异基因KEGG功能分析(kegg通路 map04626,map04075)显示,共注释3160个差异基因,映射到130条代谢通路,其中植物激素信号转导与植物病原互作包含的差异基因数量最多,分别达到118和113条,而显著富集的通路还包括淀粉和蔗糖代谢,糖酵解以及苯丙烷生物合成.这些通路在三叶木通性别分化发挥了重要作用.
2.2.6 三叶木通性别相关基因q-PCR验证
三叶木通的差异基因聚类分析以及GO富集分析均表明,Unigenes主要映射在细胞分裂素和丙酮酸代谢相关通路上,因此随机选择9个与此相关的差异基因进行q-PCR验证.结果显示这些基因均在雄花中大量表达,差异性极强(如图8~9所示)t检验表明具有统计学意义,表明这些基因可能在雄花的发育过程中起着重要作用.

注:**代表差异性极显著,P<0.01
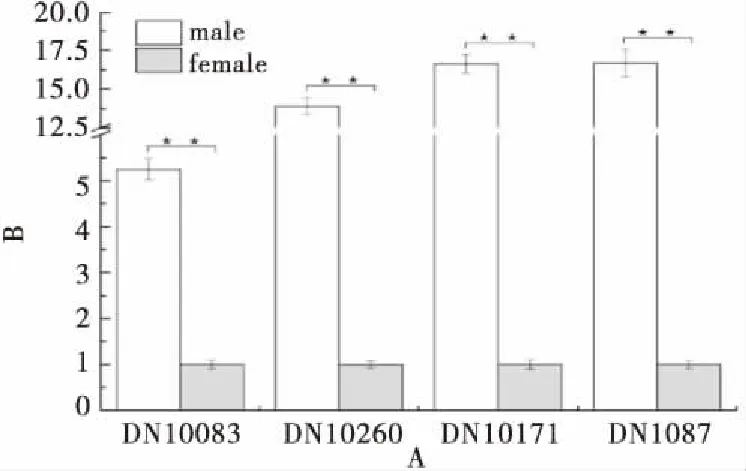
注:**代表差异性极显著,P<0.01
3 讨 论
植物性别分化是植物生长发育过程中由性别决定基因、环境因素和激素共同调控的.现有的研究表明,在部分植物中存在性染色体.日本京都大学课题组在美味猕猴桃中发现Y染色体上的特异性基因ShyGirl能够抑制心皮的发育[16],而另外一种特异性基因FriendlyBoy则能够维持雄花的发育[17].在黄瓜性别的研究中发现,自然条件下比人工培育条件下的雌花更少,推测可能是黄瓜体内的miRNA受到环境的影响选择性的使心皮败育,同时在人工培育条件下施加外源激素乙烯能够有效促使黄瓜雌花开放[18].在对杨树染色体分析后,科学家发现XIX染色体上出现了一部分Y染色体连锁的能够产生siRNA阻断对应基因的表达,从而抑制雌花生成[19].目前,对植物性别决定的研究主要还处于利用分子标记寻找特定的性别遗传基因,对于性染色体的划分以及具体作用还有一定的争议,虽然近年来分离鉴定了许多microRNA,发现甲基化能够对植物的性别决定产生影响,但性别分化的机制仍不清晰.
三叶木通的差异基因GO分析表明,三叶木通的雌雄花芽的差异基因主要在细胞组成上,与雌雄花结构差异相关.差异基因的KEGG富集分析表明,三叶木通雌雄花芽的差异基因主要富集于植物信号转导,结合前人的分析研究推断,植物激素在三叶木通性别分化的过程中起了关键的调节作用.而Unigenes的聚类分析表明丙酮酸、细胞分裂素和油菜素内酯在性别分化过程中扮演了重要的角色,参与了多种代谢过程(kegg通路map04075),细胞分裂素能够与其他激素综合调控性别分化.油菜素内酯在植物体内广泛存在,通过对油菜素内酯物的调节,能够影响花器官形成的生理过程,进而影响性别分化[20].丙酮酸参与的代谢调控网络非常复杂(如图10所示),其中在生物体内的物质代谢中起着关键桥梁作用,在糖酵解的途径中氧化生成乙酰CoA,参与三羧酸循环,从而实现生物体内三大营养物质的互相转化.而丙酮酸代谢出现问题,可能会影响绒毡层发育以及减数分裂,进而对生殖过程以及性别分化产生影响[21].

图10 丙酮酸部分代谢通路
GO分析以及qRT-PCR的验证发现,三叶木通雌雄花芽的差异基因囊括了几乎已公布的三叶木通MADS-BOX基因家族中的B类以及C类基因,这与花发育模型理论中,B类以及C类基因决定雄蕊和心皮的发育[22]是一致的,推测在植物性别分化的过程中,MADS-box家族中的基因除了直接参与花器官形态建成之外,可能也在早期参与了性别分化调控的过程.pathway的分析表明,激素在雌雄花芽的发育也起到了不可或缺的关键性作用,通过进一步检索发现,在激素相关的通路中,差异基因更多的富集在细胞分裂素相关的通路上,油菜素内酯和丙酮酸也发挥了重要调控作用.已有研究现性别分化是一个由多个因素共同调控决定的复杂的生理过程,三叶木通的性别分化机制,还有待于进一步研究.
4 结 论
本实验通过转录组测序组装,在雌雄花芽各3个转录本中一共获得了17 439个Unigenes,平均长度860 bp,N50为1 471,Q20大于98%,Q30大于93%,大部分Unigenes长度位介于200~2 000 bp之间,表明三叶木通转录组序列组装质量良好.通过实时荧光定量PCR验证,结果表明转录组数据真实可靠.将组装好的通过对比公共数据库发现,三叶木通在NR数据库比对的序列数量最多有14 327条,但在KEGG的注释只有10 017条,比例仅为57.44%.差异基因以及综合数据库的对比分析表明,三叶木通已公布的MADS-box基因均属于差异基因,同时KEGG聚类分析表明细胞分裂素和油菜素内酯的信号通路是差异基因富集较多的通路,在雄性花芽的发育过程中起着正调控作用,为后续对三叶木通的性别分化机制研究奠定了基础并提供了方向.
猜你喜欢
动物医学进展(2024年4期)2024-04-10 01:50:04
今日畜牧兽医(2022年10期)2022-12-23 06:22:28
心电与循环(2020年1期)2020-02-27 07:48:24
基层中医药(2018年2期)2018-05-31 08:45:08
Coco薇(2015年12期)2015-12-10 02:40:50
中国民族民间医药·下半月(2014年5期)2014-12-02 05:15:58
湖北农业科学(2014年3期)2014-07-21 10:25:00
食品科学(2013年22期)2013-03-11 18:29:16
食品科学(2013年17期)2013-03-11 18:27:07
中国现代中药(2011年9期)2011-09-26 08:42:53
