
龙须菜血红素加氧酶138位亮氨酸突变对藻红胆素合成的影响❋
2021-07-07 03:00黄小云臧晓南尚孟慧徐晓婷
中国海洋大学学报(自然科学版) 2021年8期
李 瑞, 黄小云, 臧晓南, 尚孟慧, 毕 莹, 徐晓婷
(中国海洋大学海洋生命科学院,海洋生物遗传学与育种教育部重点实验室, 山东 青岛 266003)
龙须菜(Gracilariopsislemaneiformis)属于红藻门(Rhodophyta),江蓠目 (Gracilariales),江蓠科(Gracilariaceae),是一种生长极其迅速的大型经济海藻[1],藻体琼脂含量高达16%~28%,在食品、医药、化工等领域都具有广泛应用[2-3]。

(Gracilaria salicornia(YP_009019632.1),Schizymenia dubyi (YP_009295967.1),Sebdenia flabellata(YP_009296171.1),Schimmelmannia schousboei(YP_009295765.1),Rhodymenia pseudopalmata(YP_009293614.1),Chondrus crispus(YP_007627407.1)。图中方框(Ⅰ~Ⅵ)表示保守结构域,深色区域为保守氨基酸。箭头所指为HO(M) 的突变位点。The box (Ⅰ~Ⅵ) in theFigure represents the conserved domain, and the dark region is the conserved amino acid. The arrow indicates the mutation site of HO(M).)
藻红蛋白(Phycoerythrin)是龙须菜中主要的藻胆蛋白,其位于藻胆体外围,能直接吸收光能,进而作为天线色素参与光合作用。而且,藻红蛋白是天然色素,可作为天然着色剂、营养保健剂等广泛应用于食品及营养研究领域[4-7]。藻红蛋白还因为具有荧光特性,常用作免疫化学和生物医学中的荧光标签[8-10]。天然藻红蛋白的荧光发射特征峰约在580 nm左右[11],其荧光发色团为开链的四吡咯结构[12],荧光发色团包括藻红胆素(Phycoerythrobilin,PEB)[13]和藻尿胆素(Phycourobilin,PUB)2种[14]。一般认为藻尿胆素的功能是传递能量[15],直接发射荧光的是藻红胆素。藻红胆素通过与脱辅基藻红蛋白上的半胱氨酸Cys结合使藻红蛋白表现出光学特性。
藻红胆素的合成起始于血红素,血红素加氧酶(Heme oxygenase, HO)将血红素氧化成第一个中间代谢产物——胆绿素(BV)IXα,该步骤为藻红胆素合成的限速步骤[16]。铁氧还蛋白依赖性色素还原酶(FDRBs)可作用于胆绿素(BV)IXα的特殊位点将其还原为藻红胆素[17-19]。(BV)IXα被还原为藻红胆素有两种代谢途径,第一种是PebS催化一个四电子还原反应直接将(BV)IXα转化成3Z-PEB;第二个途径分两步:首先(BV)IXα在PebA的催化下获得双电子被还原为15,16-DHBV;然后15,16-DHBV在PebB的催化下再获得两个电子,转化为3Z-PEB[20-22]。藻红胆素的合成是形成有光学活性藻红蛋白的重要环节,而血红素加氧酶是催化血红素降解为胆绿素这一限速步骤的关键酶类[23],在藻红胆素的合成过程中发挥至关重要的作用。
近年来关于藻红蛋白的研究报道越来越多,但大部分集中于天然藻红蛋白的分离纯化及脱辅基蛋白亚基的功能研究及表达[24-27],对于藻红胆素合成元件的来源及合成途径的研究多集中于蓝藻[28-29],而对于红藻的研究较少。本实验室前期获得了龙须菜中的血红素加氧酶基因ho-1(GenBank accession number: MH025903)[30],本研究在对ho-1基因序列进行分析后,预测了血红素(Heme)的结合位点,为了进一步验证这些位点对藻红胆素合成的作用,我们拟选取其中一个位点进行点突变,而且为了研究该突变在藻红胆素合成的两条途径中的作用,我们将突变后的ho(M)基因分别与pebS和pebB/pebA组合表达,为藻红蛋白的重组表达奠定基础。
1 材料与方法
1.1 龙须菜RNA的提取和cDNA的合成
使用RNA提取试剂盒(E.Z.N.A.®Plant RNA Kit, CAT NO: R6827, America)提取龙须菜RNA,将龙须菜RNA进行反转录,获得cDNA。置于-20 ℃环境下保存。
1.2 藻红胆素血红素加氧化酶基因ho-1的克隆
根据本实验室前期获得的龙须菜血红素加氧酶ho-1(GenBank accession number: MH025903)的序列,设计引物:
F1GGATCCATGTCTACTAATTTAGCTCAACA
R1GAGCTCTTAAGTAAATTTTTTTCGCAAACTAC
下划线所标出的序列分别为限制性酶切位点BamH I和SacI。
以龙须菜cDNA为模板,以F1和R1为引物扩增得到ho-1的序列,连接T载体获得pEASY-T1-ho-1。
测序验证序列的准确性,并对序列进行分析。应用DNAMAN[31]软件预测基因所对应的氨基酸序列;运用ExPASy(https://web.expasy.org/protparam/)在线分析软件分析蛋白质基本性质,推导预测其分子量;通过BlastP(http://www.ncbi.nlm.nih.gov/BLAST/)对氨基酸序列进行同源性比对分析;依据同源序列,应用DNAMAN进行多重序列比对获得氨基酸序列保守结构和保守氨基酸;应用SWISS-MODEL(https://swissmodel.expasy.org/)[32]对蛋白质的三级结构进行分析,预测可能的关键活性位点。
1.3 ho-1基因的点突变
根据活性位点的预测和序列分析结果,我们预测第413位碱基对应的氨基酸位点可能为血红素的关键结合位点,因此根据ho-1的序列,采用定点突变的方式,在ho-1基因中进行了一个碱基突变,将第413位碱基由T突变成C,为方便后续研究该基因称作ho(M);设计定点突变引物,扩增得到ho(M)基因。
根据ho(M)的突变位点及ho-1的序列,设计引物:
ho(M)-F1CAAAAAAAATTGCACAAACAGCTATGAATTT
ho(M)-R1 AAATTTGACCTCCCGATAAGTCACCCA
下划线所标出的位点为定点突变的位点。
以pEASY-T1-ho-1的序列为模板,扩增得到线性化的pEASY-T1-ho(M)的序列。利用Blunting Kination Ligation(BKL)Kit (TaKaRa, Code No. 6127A) 自连获得质粒pEASY-T1-ho(M)。
1.4 表达载体pET-ho-1、pET-ho-1-pebS、pET-ho-1-pebA-pebB、pET-ho(M)、pET-ho(M)-pebS、pET-ho(M)-pebA-pebB的构建
需要构建的表达载体如表1所示。pebS和pebA(GenBank:MH715937)-pebB(GenBank: MH725905)[33]片段均为本实验室前期从龙须菜中克隆得到的,且已构建了克隆载体pEASY-T1-pebS和pEASY-T1-pebA-pebB,它们将由血红素加氧酶HO催化合成的胆绿素通过两个途径分别合成藻红胆素。

表1 构建的工程菌株与重组质粒
pEASY-T1-ho-1和pEASY-T1-ho(M) 经BamHI和SacI双酶切后,将ho-1和ho(M)基因片段分别插入pET-24a多克隆位点,连接得到表达载体pET-ho-1以及pET-ho(M)。将pEASY-T1-pebA-pebB、pET-ho-1和pET-ho(M) 载体分别经SacI和XhoI双酶切后获得pebA-pebB片段和酶切后的载体,将pebA-pebB片段插入酶切后的两个载体中,得到pET-ho-1-pebA-pebB和pET-ho(M)-pebA-pebB。以成功构建好的质粒pET-ho-1-pebA-pebB和pET-ho(M)-pebA-pebB为基础,将pEASY-T1-pebS经SacI和XhoI双酶切后把pebS插入pET-ho-1-pebA-pebB和pET-ho(M)-pebA-pebB中替代pebA-pebB基因片段,从而得到表达载体pET-ho-1-pebS和pET-ho(M)-pebS。
分别取pET-ho-1、pET-ho-1-pebS、pET-ho-1-pebA-pebB,pET-ho(M)、pET-ho(M)-pebS、pET-ho(M)-pebA-pebB质粒0.5 μL,将其加入50 μL大肠杆菌BL21感受态细胞中进行转化得到表达菌株E.coliph、E.coliphS、E.coliphAB、E.coliph(M)、E.coliph(M)S和E.coliph(M)AB。
1.5 重组菌株的诱导表达
将E.coliph、E.coliphS、E.coliphAB、E.coliph(M)、E.coliph(M)S 和E.coliph(M)AB以及空白对照E.coliBL21菌种分别接种到5 mL LB液体培养基中,向培养E.coliph、E.coliphS、E.coliphAB、E.coliph(M)、E.coliph(M)S和E.coliph(M)AB的培养基中加入5 μL浓度为100 mg/mL的硫酸卡那霉素,空白对照E.coliBL21的培养基不加抗生素。在37 ℃,200 r/min条件下,培养12 h,活化所有菌液,然后将菌液转接到100 mL LB液体培养基中,在37 ℃,200 r/min条件下扩大培养2~3 h后,向扩大培养后的重组菌液中分别加入浓度为1 mol/L的IPTG至终浓度为0.1 mmol/L。在28 ℃,200 r/min诱导时间为12 h的条件下诱导重组蛋白表达。
1.6 重组菌株的蛋白电泳检测
分别取诱导表达后的E.coliph、E.coliphS、E.coliphAB、E.coliph(M)、E.coliph(M)S和E.coliph(M)AB以及空白对照E.coliBL21七个全菌菌液2 mL,10 000 r/min离心1 min,弃上清,用200 μL浓度为0.01 mol/L的PBS重悬菌体沉淀,加入50 μL 5×Loading Buffer混合,于100 ℃沸水中煮沸5 min,进行SDS-PAGE检测,分离胶浓度为15%,浓缩胶浓度为5%,上样量为20 μL,电压170 V,电泳时间约为1 h。
1.7 重组菌株的荧光发射光谱检测
收集诱导后的菌体沉淀,用0.9%的生理盐水洗涤菌体沉淀两次,加入3 mL浓度0.01 mol/L的PBS重悬菌体沉淀,使用超声破碎仪超声破碎6 min,破碎5 s间隔2.5 s;离心取上清液使用HITACHIFI-4600型荧光光谱仪进行荧光发射光谱分析。荧光光谱扫描速度1 200 nm/min,藻红蛋白检测时激发波长为420 nm,狭缝宽度5.0 nm。
2 结果与分析
2.1 ho-1和ho(M)基因的克隆与序列分析
以龙须菜cDNA为模板,扩增得到一段长度约700 bp的条带,测序结果显示其实际大小为696 bp,与序列MH025903一致,确定其为ho-1基因。
用DNAMAN软件分析ho-1基因序列,cDNA编码框序列长693 bp,其起始密码子为ATG,终止密码子为TAA。ho-1基因开放阅读框编码231个氨基酸,其中含有107个非极性氨基酸(A、V、L、I、F、W、Y、M、P),69个极性氨基酸(G、N、Q、S、T、C),23个酸性氨基酸(D、E)和32个碱性氨基酸(K、H、R)。ExPASy预测HO-1分子量为26.3 kDa,等电点为9.30。
突变序列ho(M)只在第413位碱基由T突变成C,其他位置ho-1和ho(M) 的序列完全一致(见图1)。

图1 ho-1、ho(M)的cDNA核酸序列比对图
多序列比对显示龙须菜的血红素加氧酶HO-1的氨基酸序列与其他藻类的同源序列比对的相似性为85.77%,说明血红素加氧酶HO-1在藻类中比较保守。其氨基酸序列存在6个保守结构区域,如图2所示,分别是MSTNLAQ,LREGTTK,HSMAENVSFVKSFLGGVVD,ANLFFVY,NQPELLIAHAYTRYMGDLSGGQILKKIA,FQELNSN;以及多个保守氨基酸残基,其中位于115~142位的结构域保守性最强,推测其在保持血红素氧化酶的功能上具有重要作用。
对ho-1序列预测获得的氨基酸序列进行BlastP分析结果显示,其属于HemeO superfamily超级家族且氨基酸序列上存在17个可能的血红素(Heme)结合位点(见图3),分别是R10、H17、A20、E21、V26、L30、Y125、T126、R127、S133、G134、L138、K169、R173、F197、N200、F204。118~145位存在一段HemeO保守结构域的扭结螺旋结构。推测这些位点能够与血红素结合,进而催化血红素转化为胆绿素,发挥血红素氧化酶的功能。

(▲所示为可能的血红素(Heme)结合位点。118~145位长条显示为一段HemeO保守结构域的扭结螺旋结构。Possible heme binding sites are shown as ▲. The 118~145 band shows a spiral structure in the HemeO conservative domain. )
利用SWISS-MODEL对HO-1的三级结构进行分析,参照集胞藻Synechocystissp. PCC6803的血红素氧化酶heme oxygenase-1的结构建模结果如图4所示,HO-1内部会形成一个血红素结合口袋(见图4A),有18个可能的活性位点与Heme连接(见图4B),包括R10、H17、A20、V26、L30、Y125、T126、M129、G130、S133、G134、I137、L138、K169、R173、F197、N200、F204。其中有15个位点R10、H17、A20、V26、L30、Y125、T126、S133、G134、L138、K169、R173、F197、N200、F204与BlastP预测的血红素结合位点一致。
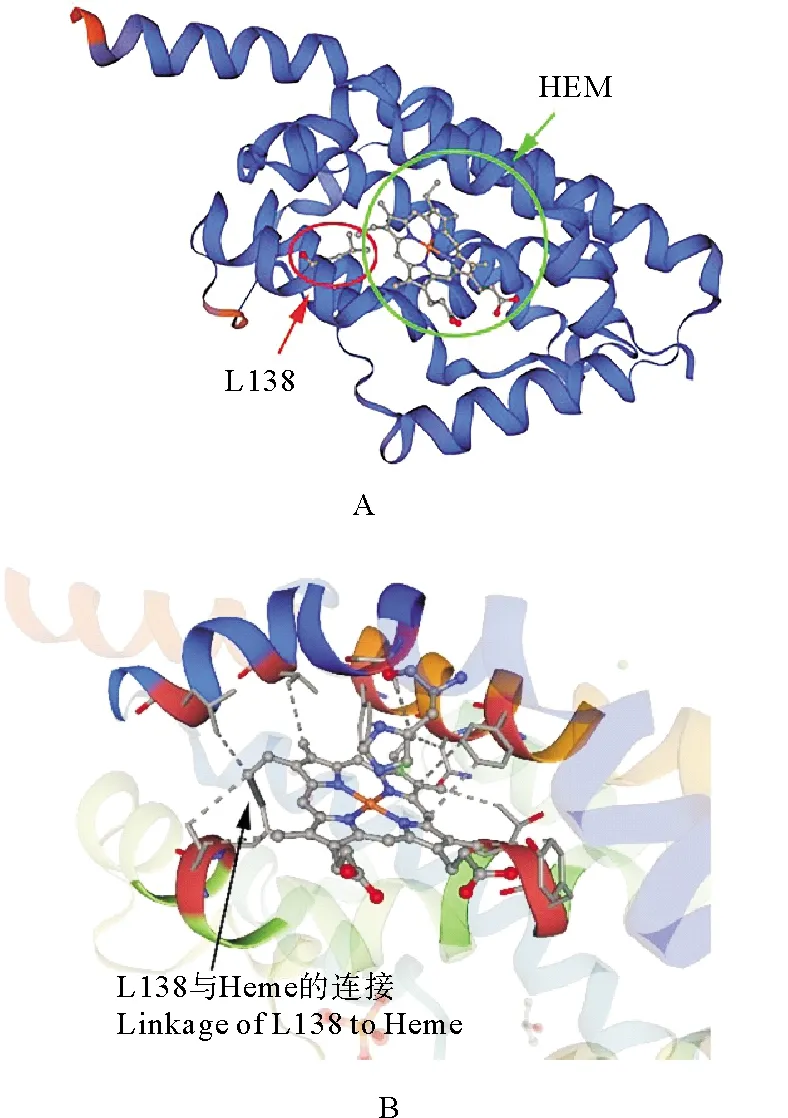
(A.预测的HO-1的三维结构图,其中HEM为血红素,L138为138位亮氨酸;B.预测的血红素与HO-1的连接,其中箭头所示为L138与血红素的连接。A. 3D-structure diagram of predicted HO-1, where HEM is heme and L138 is leucine at position 138; B. Predicted heme to HO-1 linkage, where arrow indicates L138 to heme linkage.)
本研究引入的突变位于ho-1基因的第413位,由碱基T突变成C,对应编码的第138位氨基酸由亮氨酸(L,非极性氨基酸)突变为丝氨酸(S,极性氨基酸)。该位点经BlastP和SWISS-MODEL两种程序都预测是血红素的结合位点,其位于118~145位HemeO保守结构域的扭结螺旋结构内,而且该位点还在115~142位的高度保守的结构域内,因此接下来我们通过重组表达和荧光活性观察来确定该位点在藻红胆素合成中的作用。
2.2 HO-1和HO(M)蛋白的重组表达
将重组菌株E.coliph、E.coliph(M)、E.coliphS、E.coliph(M)S、E.coliphAB及E.coliph(M)AB经诱导表达后进行蛋白电泳检测,均在25~30 kDa间检测到蛋白条带,与HO-1蛋白预测的大小26.3 kDa一致,而空白对照没有此蛋白条带,表明HO-1蛋白及其突变蛋白HO(M)都可以在大肠杆菌E.coli中正常表达(见图5)。
2.3 重组表达菌株的荧光发射光谱分析
图6A为空白对照E.coliBL21与重组菌株E.coliph及E.coliph(M)的荧光发射光谱图,重组菌株E.coliph在595 nm附近检测到微弱的荧光峰,该位置近似于藻红胆素的荧光特征峰;而E.coliph(M)及E.coliBL21均未检测到该特征峰,初步表明ho-1基因的第413位碱基突变后,其催化胆绿素合成的功能受到影响。
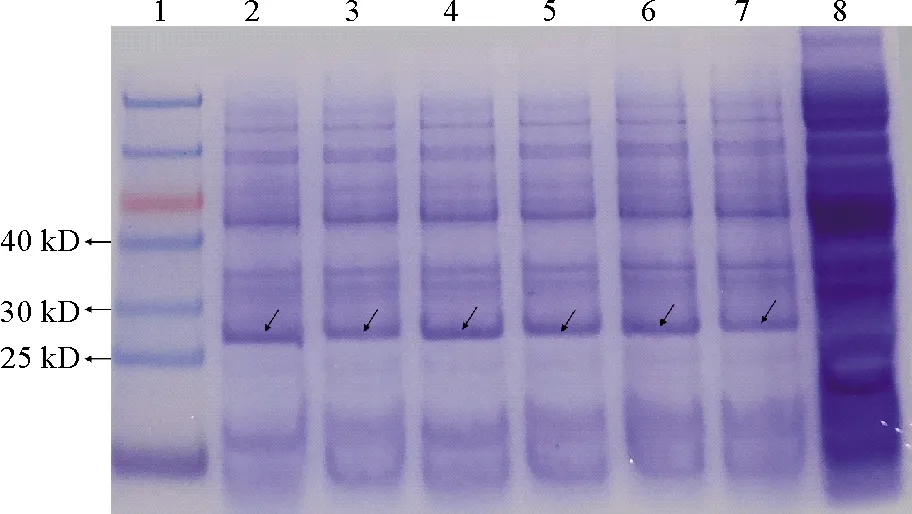
(泳道1为Marker (14~100 kD),泳道2~7分别为E. coli ph、E. coli ph(M)、E. coli phS、E. coli ph(M)S、E. coli phAB及E. coli ph(M)AB;泳道8为空白对照E. coli BL21,箭头表示HO-1和HO(M) 蛋白。Lane 1 is Marker (14~100 kD), lane 2~7 are respectively E. coli ph, E. coli ph(M), E. coli phS, E. coli ph(M)S, E. coli phAB, E. coli ph(M)AB. Lane 8 is the blank control E. coli BL21.The arrows represent HO-1 and HO(M) proteins.)
在此基础上,继续研究重组菌株E.coliphS、E.coliph(M)S、E.coliphAB及E.coliph(M)AB的荧光活性(见图6B、C),发现重组菌株E.coliphS和E.coliphAB在595 nm附近有明显的藻红胆素的荧光特征峰,表明HO-1能够催化血红素合成胆绿素,胆绿素在铁氧还蛋白依赖性色素还原酶PebA和PebB或者PebS两条途径下都可以催化合成藻红胆素;而在突变后的E.coliph(M)S及E.coliph(M)AB与E.coliph(M)及空白对照E.coliBL21一样无任何荧光峰出现,进一步表明ho(M)由碱基T突变成C的第413位碱基位点十分关键,其对应编码的第138位亮氨酸对于酶的活性至关重要,该位点的突变使得PebS和PebB/A两条催化途径均无法合成藻红胆素,直接影响藻红胆素的合成。
比较重组菌株E.coliph、E.coliphS、E.coliphAB的荧光峰值(见图6D)可见,E.coliph的荧光峰值略低于E.coliphS、E.coliphAB,可能是因为在HO-1的作用下,只能由血红素合成胆绿素,没有完整的藻红胆素合成,因而荧光值较低;在重组菌株E.coliphS、E.coliphAB中,于630 nm处也检测到微弱的荧光峰,推测此荧光峰可能是由在藻红胆素合成过程中所生成的一些中间体产生的。
3 讨论
对于藻类血红素加氧酶的研究始于单细胞红藻Cyanidiumcaldarium[34-35], 研究发现,该藻类中的血红素加氧酶是由核基因编码,在细胞质核糖体上合成的可溶性酶;在红藻C.caldarium的提取物中已经检测到将原血红素转化为胆绿素IXα和将胆绿素IXα转化为藻红胆素及藻蓝胆素的酶反应,并对其进行了表征[23, 36-40]。之后,Reith等[41]构建了红藻紫菜Porphyrapurpurea叶绿体基因组高分辨率基因图谱,并从中获得了编码血红素加氧酶的基因pbsA,这是首次在叶绿体基因组上检测到该基因。1996年,在蓝藻集胞藻Synechocystissp. PCC6803菌株中鉴定出两个不同的血红素加氧酶基因,命名为ho[42];后来,在其提取物中检测到了藻胆素生物合成的反应,这是体外血红素加氧酶活性和原核细胞提取物中胆绿素IX向藻胆素转化的首次报道[43]。1998年,在大肠杆菌中克隆并表达了来自集胞藻PCC6803的ho1,在其细胞提取物中产生血红素加氧酶活性[44]。随后,Glazer 和 Tooley 等[45]将集胞藻Synechocystissp. PCC6803的藻蓝蛋白合成相关基因 (cpcA,cpcE,cpcF,hox1,pcyA) 导入大肠杆菌表达细胞中,成功实现了具有光学活性藻蓝蛋白α亚基的合成。
本研究以龙须菜作为材料,对克隆得到的龙须菜血红素加氧酶ho-1基因的序列进行分析,发现其第138位氨基酸经BlastP和SWISS-MODEL两个软件都预测是血红素的结合位点,其位于118~145位HemeO保守结构域的扭结螺旋结构内,而且该位点还在115~142位的高度保守的结构域内。为了验证该位点的功能,本研究对ho-1基因内的413位碱基由T突变为C,对应的138位氨基酸由亮氨酸(L,非极性氨基酸)突变为丝氨酸(S,极性氨基酸),然后构建ho-1及其突变基因ho(M)的异源重组表达菌株,并在诱导温度28 ℃,IPTG工作浓度为0.1 mmol/L,诱导时间为12 h的条件下诱导重组蛋白表达;蛋白电泳结果显示相关重组表达蛋白已正常表达。荧光发射光谱显示,重组菌株E.coliphAB和E.coliphS在595 nm处可以检测到荧光发射峰,表明已生成具有正常光学活性的藻红胆素,即HO-1能够正常催化血红素合成胆绿素,且合成的胆绿素可以再由两条途径合成藻红胆素,一条是由铁氧还蛋白依赖性色素还原酶PebS直接催化合成PEB;第二条是由铁氧还蛋白依赖性色素还原酶PebA和 PebB依次催化合成 PEB。而突变菌株E.coliph(M)、E.coliph(M)S、E.coliph(M)AB与E.coliBL21一样无任何特征峰出现,表明由于ho(M) 第413位碱基位点的突变,使得血红素加氧酶HO失活,不能够合成胆绿素,进而使得PebS和PebB/A两条催化途径均无法合成藻红胆素,说明第138位的亮氨酸位点在藻红胆素合成过程中十分关键。

(A. 重组菌株E. coli ph、E. coli ph(M)及空白对照E. coli BL21的荧光发射图谱;B. 重组菌株E. coli phS、E. coli ph(M)S及空白对照E. coli BL21的荧光发射图谱;C. 重组菌株E. coli phAB、E. coli ph(M)AB及空白对照E. coli BL21的荧光发射图谱;D. 重组菌株E. coli ph、E. coli phS、E. coli phAB及空白对照E. coli BL21的荧光发射图谱重组菌株。A. Fluorescence emission spectrum of recombinant strains E. coli ph, E. coli ph(M) and E. coli BL21; B. Fluorescence emission spectrum of recombinant strains E. coli phS, E. coli ph(M)S and E. coli BL21; C. Fluorescence emission spectrum of recombinant strains E. coli phAB, E. coli ph(M)AB and E. coli BL21; D. Fluorescence emission spectrum of recombinant strains E. coli ph, E. coli phS, E. coli phAB and E. coli BL21.)
与天然藻红蛋白(EX:496 nm,EM:580 nm)相比,重组藻红蛋白的荧光激发波长(EX:420 nm)发生了蓝移,而荧光发射波长(EM:595 nm)发生了红移,这可能是由于体外重组的藻红胆素与天然藻红胆素在结构及化学组成上有所差异,因此其光谱性质也存在不同。对于天然的R-藻红蛋白来说,其在580 nm处的单个荧光最大值是从PUB到PEB发色团的荧光共振能量转移(FRET)的结果[11],而在重组藻红蛋白中由于PUB的缺失,可能并不存在这种能量转移。
此外,重组菌株E.coliphAB和E.coliphS在630 nm处还检测到一个荧光峰(见图6D),推测该荧光发射峰可能是藻红胆素合成过程中的中间体。藻红胆素合成过程首先是由血红素加氧酶HO-1催化血红素形成胆绿素,进而在铁氧还蛋白依赖性色素还原酶催化下生成藻红胆素。在体外重组表达过程中,各种酶的表达水平可能会不均衡,从而导致不完全的中间体产生。
目前,多种血红素加氧酶已被人们获得:如动物中的微粒体血红素加氧酶[46],某些红藻、蓝藻等藻类及高等植物中的血红素加氧酶[47]等,本文对龙须菜血红素加氧酶基因ho-1的克隆及研究进一步加深了对藻类中HO的了解,说明了HO在结构上的保守性及其在藻红胆素正常合成中的关键作用,为之后研究藻红蛋白的结构、合成途径、光谱性质及聚集状态奠定基础;其强大的光学活性和良好的应用前景也为后续重组藻红蛋白的研究带来信心。
猜你喜欢
分子催化(2022年1期)2022-11-02
草地学报(2022年3期)2022-03-28
中西医结合肝病杂志(2021年7期)2021-11-30
今日农业(2021年11期)2021-11-27
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
中国食用菌(2020年11期)2021-01-18
电脑知识与技术(2018年19期)2018-11-01
医学研究杂志(2015年9期)2015-07-01
食品工业科技(2014年13期)2014-03-13
