
柱色谱分离-分子筛络合洗脱过程中正构烷烃单体碳同位素分馏研究
2021-07-06 06:04董浩伟赵佳玉曾凡刚谢曼曼尚文郁王淑贤孙青
岩矿测试 2021年3期
董浩伟, 赵佳玉, 曾凡刚, 谢曼曼, 尚文郁, 王淑贤*, 孙青*
(1.国家地质实验测试中心,北京100037;2.中国科学院地质与地球物理研究所新生代地质与环境重点实验室, 北京100029;3.中国科学院地球科学研究院,北京100029;4.中国科学院大学,北京100049;5.中国人民大学环境学院,北京100872)
来源于生物体的正构烷烃能够较稳定地保存于地质体中[1-2]。不同链长的正构烷烃能够大致“标记”其物质来源。短链正构烷烃(nC15~nC21)主要来源于水体中的藻类和微生物[3];中链正构烷烃(nC23~nC25)主要来源于挺水植物、浮水植物和苔藓[4];长链正构烷烃(nC27~nC35)则主要来源于陆生高等植物[5]。不同链长的正构烷烃单体碳同位素被广泛应用于示踪沉积物物源[6-11],重建古植被[12]、古气候[13-17]、古环境[18-22]和古地形[23]等。例如,Rommerskirche等[12]对比了全新世非洲大陆边缘沉积物的孢粉和正构烷烃单体碳同位素数据,指出正构烷烃单体碳同位素可以有效地重建植被演变历史。Yamada等[18]分析了日本海洋沉积物的长链正构烷烃单体碳同位素(n-δ13C29、n-δ13C31),重建了日本南部距今8.5万年以来的古环境演变历史。饶志国等[24-25]对比研究了中国东部表土总有机质碳同位素和长链正构烷烃碳同位素,指出表土总有机质碳同位素和陆生高等植物来源的长链正构烷烃碳同位素同等有效地记录了来源植被中C3/C4植物的相对生物量贡献。
利用气相色谱-气体同位素质谱仪(GC-C-IRMS)分析正构烷烃单体碳同位素之前,需要对饱和烃样品中正构烷烃和异构烷烃进行预分离、富集处理,目的是减少未分峰和共流出的干扰,并满足同位素质谱仪的检出限,提高分析的精密度和准确度[26-32]。目前,分离富集样品中正构烷烃的常用方法有硫脲络合法、尿素络合法、高效液相色谱法、柱色谱法、气相色谱法和5Å分子筛法等[33-36]。其中5Å分子筛法包括氢氟酸酸解法和混合溶剂洗脱法。氢氟酸酸解法使用氢氟酸破坏分子筛结构释放正构烷烃,但氢氟酸毒性较大、不安全。混合溶剂洗脱法采用正戊烷、环己烷混合溶剂进行洗脱,需要经过柱色谱、5Å分子筛络合、环己烷-正戊烷混合溶剂多次洗脱过程,前处理流程复杂,影响因素多,回收率不稳定。前处理过程中正构烷烃单体碳同位素是否发生分馏,是正构烷烃单体碳同位素高精度分析的关键。
已有研究表明正构烷烃回收率约50%或以上时,5Å分子筛法不会造成正构烷烃单体同位素分馏。例如,Tolosa等[34]研究了样品经5Å分子筛络合、氢氟酸溶解后得到的正构烷烃,发现正构烷烃回收率平均值为53%时碳同位素没有发生明显分馏。张逐月等[28]应用5Å分子筛络合、环己烷-正戊烷混合溶剂洗脱分离富集正构烷烃,正构烷烃回收率平均值为59%,正构烷烃单体碳同位素分析精度优于0.38‰,没有发生明显同位素分馏。Grice等[33]发现石油样品经5Å分子筛络合混合溶剂洗脱,正构烷烃回收率基本在90%以上,未观察到前处理造成正构烷烃的碳同位素分馏。上述研究表明,当正构烷烃回收率较高时,前处理过程不会对单体同位素值造成影响。然而,当回收率较低时,前处理过程是否会引起碳同位素分馏,以及前处理中各步骤是否会导致正构烷烃单体同位素变化仍有待开展深入研究。本文以正构烷烃混合溶液为对象,分析了经柱色谱、5Å分子筛络合、混合溶剂洗脱前后的正构烷烃单体碳同位素比值,讨论了柱色谱分离前后、5Å分子筛不完全络合前后、两次洗脱处理前后、正构烷烃不同洗脱回收率时的正构烷烃单体碳同位素变化特点,着重探讨正构烷烃低回收率时,前处理过程是否会引起正构烷烃单体碳同位素分馏。
1 实验部分
1.1 仪器及工作条件
GC-2010气相色谱仪(日本岛津公司),柱箱初始温度为60℃,以10℃/min程序升温至250℃,再以6℃/min程序升温至320℃,恒温7min。
TraceGC ULTRA-GC ISOLINK接口-MAT253质谱仪(美国ThermoFisher公司)。柱箱初始温度为70℃,以4℃/min程序升温至320℃,保持10min。氧化炉温度1000℃。
EG20A plus电热板(北京莱伯泰科仪器有限公司);XMTA-C9000马弗炉(天津市泰斯特仪器有限公司)。
载气:高纯氮气和高纯氦气(99.999%,北京市北温气体制造厂)。
1.2 材料和主要试剂
硅胶:500mg/3mL SPE硅胶柱(美国Waters公司)。
接收瓶:60mL透明收集瓶(美国ThermoFisher公司)。
正戊烷、正己烷、环己烷、二氯甲烷:色谱纯(美国ThermoFisher公司)。
工作标准:nC7~nC40正构烷烃混合工作标准(美国Sigma公司)。
正构烷烃:nC15、nC17、nC21、nC22、nC24、nC26(色谱纯,美国Sigma公司);nC18、nC28、nC32、nC36(分析纯,上海安谱公司)。
1.3 实验方法
1.3.1模拟样品配制
模拟样品是由nC15、nC17、nC18、nC21、nC22、nC24、nC26、nC28、nC32、nC36正构烷烃配制而成,单体浓度约为500μg/mL。
1.3.2柱色谱分离
选用500mg/3mL SPE硅胶柱,经2mL正戊烷淋洗获得饱和烃组分。
1.3.35Å分子筛络合与分析
5Å分子筛在使用前需在450℃连续活化24h,活化后放置于干燥器中保存。
样品置于60mL接收瓶中,加入适量活化好的分子筛和环己烷。将接收瓶密封,80℃加热24h。加热结束后待容器冷却至室温,收集上清液并浓缩。用环己烷冲洗分子筛4~5遍,并将冲洗溶剂与前次收集的上清液合并浓缩。利用气相色谱分析其中正构烷烃含量,GC-C-IRMS分析正构烷烃单体碳同位素值。
1.3.45Å分子筛洗脱与分析
在装有5Å分子筛的ASE接收瓶中,加入体积比为91∶9的正戊烷和环己烷混合溶剂。密封加热(85℃,8h),第一次洗脱后将此次洗脱液浓缩、定容,用气相色谱和GC-IRMS分别测定正构烷烃含量及其单体碳同位素值;第二次洗脱后将此次洗脱液浓缩、定容,同样用气相色谱和GC-IRMS分别测定正构烷烃含量和单体碳同位素值。
本文涉及的未络合率、柱色谱回收率、5Å分子筛回收率定义如下:

碳同位素比值(δ13C),按如下公式计算:
式中:碳同位素比值δ13C以δ13C-VPDB表示。
2 结果与讨论
2.1 仪器稳定性检验
单体碳同位素分析中仪器状态的稳定是数据质量的保证[29]。在相同实验条件下每隔6个样品穿插分析一次正构烷烃工作标准单体碳同位素,计算其标准偏差(SD),进行仪器稳定性检验。正构烷烃工作标准单体碳同位素分析精度优于0.2‰(表1),表明仪器状态稳定[26-29],正构烷烃δ13C测定结果可靠。

表1 工作标准的δ13C值
2.2 柱色谱分离前后单体碳同位素比值对比
柱色谱分离是由液固萃取柱和液相色谱技术相结合发展而来的分离、净化方法,它是生物标志化合物[37]和有机污染物[38-39]单体同位素分析常用的处理方法。Benbow等[40]认为柱色谱处理会引起水溶性有机化合物碳同位素分馏,但柱色谱处理是否会导致脂溶性正构烷烃碳同位素分馏有待进一步研究。
本文利用柱色谱处理配制的模拟样品,柱色谱处理前后的正构烷烃回收率为86%~93%。柱色谱处理后,多数正构烷烃单体碳同位素比值与模拟样品相差-0.2‰~0.2‰(图1),同位素分析精度与柱色谱分离水溶性有机化合物碳同位素分析精度[40]相似,表明柱色谱处理前后正构烷烃单体碳同位素没有发生分馏。仅n-δ13C32差值较大(-0.7‰,图1),可能是因为nC32工作标准中含有一定杂质,对碳同位素比值的测定造成了影响。

图1 过柱后单体碳同位素与模拟样品碳同位素的差值Fig.1 Difference between compound specific carbon isotope and simulate sample carbon isotope after passing through the SPE column
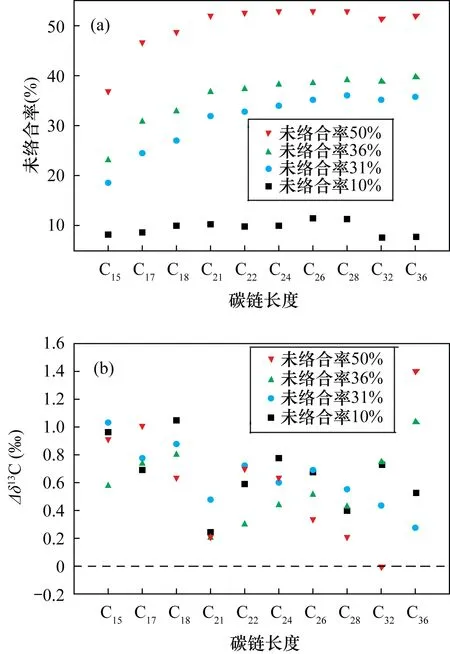
a—正构烷烃未络合率; b—正构烷烃单体碳同位素与模拟样品的差值。图2 不同未络合率正构烷烃单体碳同位素与模拟样品的差值Fig.2 Difference between carbon isotopes of n-alkanes with different uncomplexation ratios and simulate sample. (a) The uncomplexation ratio of n-alkanes; (b) The difference value between carbon isotopes of n-alkanes and simulate sample
2.3 5Å分子筛不完全络合前后单体碳同位素比值对比
实验设计了样品未被5Å分子筛完全络合的情况,试图探讨样品中正构烷烃不完全络合时单体碳同位素是否会产生明显分馏。
在络合时间、络合温度、正构烷烃含量等条件不变的情况下,通过减少5Å分子筛加入量,使得正构烷烃发生不同程度地不完全络合,未络合率为10%~50%(图2a)。其中低碳链正构烷烃的未络合率较低,高碳链正构烷烃的未络合率较高,其原因可能是低碳链正构烷烃容易被分子筛络合,或浓缩过程中更易发生挥发损失[27-28]。将未络合的正构烷烃单体碳同位素与模拟样品进行对比,发现未络合的单体碳同位素整体偏重(平均值约0.7‰,图2b),短链(nC15~nC21)正构烷烃比中长链(nC23~nC35)偏重更显著。虽然分析过程中,未络合正构烷烃的比例一般较小,但考虑到未络合部分的单体碳同位素发生了微弱的同位素分馏,建议分析过程中尽量避免正构烷烃的不完全络合。
2.4 两次洗脱处理前后单体碳同位素比值对比
由于同位素质谱仪的检出限较高,而一些环境样品中的正构烷烃含量低,为了满足分析要求,需要采用多次洗脱以提高样品中正构烷烃的回收率[27-28]。例如,陈莎莎等[27]对样品进行了三次混合溶剂洗脱,正构烷烃平均回收率提高了19%。张逐月等[28]对土壤样品进行了两次混合溶剂洗脱,也证明了两次洗脱会对正构烷烃回收率有所提高。但多次洗脱需要多次长时间的加热,可能会导致正构烷烃单体碳同位素发生分馏[27]。
本实验中,样品经过5Å分子筛完全络合后,利用混合溶剂洗脱两次,分析正构烷烃回收率及单体碳同位素比值。第一次洗脱后正构烷烃回收率为37%~90%,短链正构烷烃回收率大于50%,中链正构烷烃回收率较高(大于70%),而长链正构烷烃回收率较低(约40%)。第二次洗脱后正构烷烃回收率增加了5%~17%,其中长链正构烷烃的回收率显著提高(图3a)。
第一次洗脱后获得的正构烷烃单体碳同位素与模拟样品相差在-0.2‰~0.5‰,第二次洗脱后此差值在-0.3‰~0.2‰。两次洗脱获得的正构烷烃碳同位素比值均在分析误差之内,未发生整体的偏轻或偏重(图3b)。中短链正构烷烃单体碳同位素偏差较小,但长链n-δ13C32和n-δ13C36偏差较大,可能是因为模拟样品中含有一定杂质,对nC32、nC36碳同位素分析造成了影响。实验结果表明,两次洗脱不会导致正构烷烃单体碳同位素分馏。
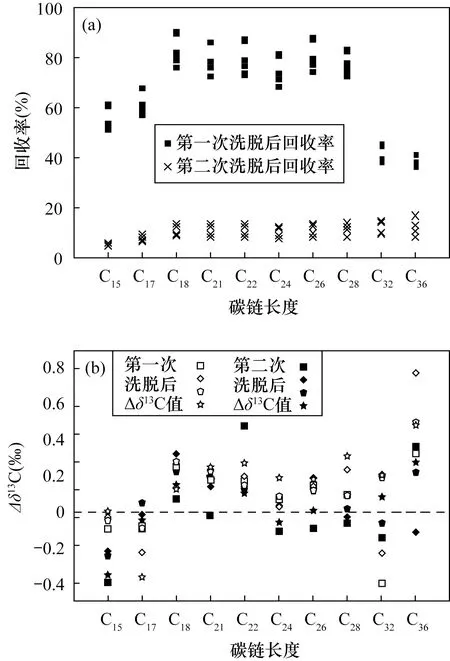
a—正构烷烃回收率; b—正构烷烃单体碳同位素与模拟样品的差值。图3 不同洗脱回收率正构烷烃单体碳同位素与模拟样品的差值Fig.3 Difference between monomer carbon isotope of n-alkanes and simulate sample with different elution recoveries. (a) The recovery rate of n-alkanes; (b) The difference value between carbon isotopes of n-alkanes and simulate sample
2.5 正构烷烃不同洗脱回收率时的碳同位素比值对比
在络合时间、络合温度等条件不变的情况下,模拟样品经过5Å分子筛处理得到不同回收率的正构烷烃。影响正构烷烃回收率的原因有:①改变分子筛加入量。例如回收率17%是减少分子筛用量、不完全络合造成的;②与氮吹浓缩有关[27-28];③冲洗分子筛时,振荡不充分导致正构烷烃不完全洗脱[28];④空气湿度的变化[41]等。由图4a可知,中短链正构烷烃回收率较高,nC32~nC36回收率较低。如nC15~nC25正构烷烃回收率为15%~80%,nC32~nC36正构烷烃回收率为10%~40%。
正构烷烃回收率及络合率不同的情况下,其碳同位素与模拟样品碳同位素的差值基本在0.3‰以内(图4b),表明回收率及络合率均不会对同位素比值造成影响。这与前人的研究结果一致[27-29,33-34]。但是前人的研究多集中在高回收率的样品,而本实验结果表明正构烷烃单体回收率低至20%,或高至90%,前处理未对正构烷烃单体碳同位素造成明显的分馏。

a—正构烷烃回收率; b—正构烷烃单体碳同位素与模拟样品的差值。图4 不同回收率正构烷烃单体碳同位素与模拟样品碳同位素差值Fig.4 Carbon isotope difference between n-alkanes monomer and simulate sample with different recoveries. (a) The recovery rate of n-alkanes; (b) The difference value between carbon isotopes of n-alkanes and simulate sample
3 结论
本研究对比了样品处理前后正构烷烃的碳同位素比值,结果表明固相萃取-5Å分子筛络合-混合溶剂洗脱前处理过程对正构烷烃单体碳同位素分馏不会造成明显影响:①固相萃取、混合溶剂两次洗脱前后正构烷烃单体碳同位素组成不存在显著差异。②该方法适用于正构烷烃回收率大于20%的样品中正构烷烃单体碳同位素比值分析。
此外,5Å分子筛不完全络合时,未络合的单体碳同位素整体偏重,可能会发生微弱的碳同位素分馏,但并未影响洗脱后的正构烷烃单体碳同位素比值,对该现象还有待进一步研究。
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10
云南化工(2021年11期)2022-01-12
云南化工(2021年10期)2021-12-21
石油石化绿色低碳(2019年6期)2019-01-14
教学考试(高考化学)(2017年3期)2017-08-08
草业学报(2016年6期)2016-09-01
中国石油大学学报(自然科学版)(2015年2期)2015-11-10
中国环境科学(2014年6期)2014-05-24
石油化工应用(2014年12期)2014-03-11
石油化工应用(2014年2期)2014-03-11
