
氟改性UiO-66固载钼基过氧化物催化氧化含硫化合物
2021-06-29 06:01张仁丽遇治权孙志超王安杰刘颖雅
高等学校化学学报 2021年6期
张仁丽,王 瑶,遇治权,2,孙志超,2,王安杰,2,刘颖雅,2
(1.大连理工大学化工学院,2.精细化工国家重点实验室,大连 116024)
我国治理环境污染的现实需要提升燃油的品质[1].加氢脱硫(HDS)是工业上脱除燃料中含硫化合物的主要方法,可以有效脱除硫醇、硫醚等含硫化合物.柴油中的含硫化合物主要为二苯并噻吩(DBT)等杂环含硫化合物,其加氢反应(还原)活性低于单环的噻吩和双环的苯并噻吩,反过来其氧化反应活性则高于单环和双环的含硫化合物.显然,通过氧化反应脱除残余的稠环含硫化合物,其效率高于加氢脱硫方法[2~5].近年来,氧化脱硫(ODS)技术因其反应条件温和、投资成本低、能够去除难以被加氢脱硫去除的噻吩类含硫化合物而被认为是目前最具前途的深度脱硫技术之一[6~8].
氧化脱硫工艺常见的多相催化体系包括负载型过渡金属氧化物如氧化钼、氧化钨、氧化钒等或者杂多酸盐,这类高氧化态过渡金属,如Mo(Ⅵ),W(Ⅵ),V(Ⅴ)和Ti(Ⅳ)等,常用于催化液相氧化反应,一般的活性顺序为Mo(Ⅵ)>W(Ⅵ)>V(Ⅴ)>Ti(Ⅳ)[9].如何选择合适的载体,在保持活性组分高活性的同时通过二者的相互作用稳定活性组分是关键.金属有机框架(MOF)是一类具有较高比表面积,易于修饰的多孔晶体材料[10,11],部分MOF骨架上的过渡金属离子配位不饱和,具有一定的Lewis酸性,在氧化脱硫反应中表现出一定的催化活性.另一方面,MOF通过化学修饰的方法可以实现活性组分在MOF 材料上的高度分散,在催化氧化脱硫中表现出优势.因此,大量氧化脱硫的研究工作围绕MOF作为催化剂或者催化载体开展.如Liu等[12]用含有联吡啶位点的Ga-MOF材料锚定了MoO2Cl2配合物,并研究了Mo@COMOC-4 在固定床反应器上的催化氧化脱硫性能,结果表明,MoO2Cl2在氧化过程中生成Mo的过氧物种是催化活性中间体.在众多MOF材料中,锆基MOF材料由于其稳定的锆氧簇单元而具有较好的化学稳定性,常用于催化反应.Zr基UiO-66具有一定的氧化脱硫活性,其催化活性来自于Zr 配位不饱和的缺陷位点形成的Lewis 酸中心[13,14].通过改性,制备含有更多缺陷位点的UiO-66能够有效提高其酸催化性能[15~17].Afzali等[18]利用UiO-66中锆氧簇上的羟基与MoO2(acac)2成键,通过Zr—O—Mo键将活性组分锚定在UiO-66骨架上,所制备的催化剂MoUiO-66用于催化含硫化合物氧化,值得一提的是,通过引入醋酸作为助催化剂,在双氧水作氧化剂的条件下,该催化体系能够在室温(25 ℃)下快速实现系列含硫化合物的氧化,如催化DBT,在最优条件下,35 min 内DBT 转化率达到97%,然而循环稳定性不佳,经4次循环反应后DBT转化率由98%降低至80%,其原因是由于钼活性组分的流失.Corma等[19]研究了Ti-MCM-41及MoOx/Al2O3催化剂在固定床反应器上的催化氧化脱硫性能,证实砜类产物吸附在亲水性的载体上使催化剂失活,而通过对载体进行硅烷化处理,构建具有一定疏水性的表面,能够有效抑制砜的吸附,从而提高催化剂的使用寿命.因此,可以认为对催化剂进行疏水改性从而对氧化脱硫产物吸附过程进行抑制是可能的.疏水改性不仅可以提高催化剂的活性,而且可以提高催化剂的循环稳定性.
本文通过改性UiO-66,引入氟基调控骨架的亲疏水性,引入氨基并通过后修饰法锚定MoO(O2)2.并将其用于DBT氧化反应,优选出最佳配体投加量,通过正交实验确定最佳反应条件,并考察了其循环稳定性.
1 实验部分
1.1 试剂与仪器
氢氧化钾、对苯二甲酸(H2BDC)、N,N-二甲基乙酰胺(DMA)、N,N-二甲基甲酰胺(DMF)、四水合钼酸铵、三氯甲烷、乙腈、无水乙醇、联苯(BP)、正庚烷及甲苯购自国药集团化学试剂有限公司;3-氟-4-甲基苯甲酸、2-氨基-对苯二甲酸(H2BDC-NH2)和叔丁基过氧化氢水溶液(TBHP,纯度70%)购于梯希爱(上海)化成工业发展有限公司;高锰酸钾、乙酸和过氧化氢水溶液(质量分数30%)均购于天津市科密欧化学试剂有限公司;水杨醛、过氧化氢异丙苯(CHP,80%异丙苯溶液)、二苯并噻吩(DBT)、苯并噻吩(BT)和4,6-二甲基苯并噻吩(4,6-DMDBT)购于上海阿拉丁生化科技有限公司.所用试剂均为分析纯.
Rigaku D/Max 2400 型X 射线衍射仪(XRD,日本理学公司,CuKα射线,λ=0.15418 nm,扫描范围5°~50°,扫描速率8°/min,电压40 kV,电流100 mA);Autosorb Ⅰ型物理吸附仪(美国康塔公司);NAVA nanoSEM 450 型场发射扫描电子显微镜(SEM,美国FEI 公司);Equinox55 型傅里叶变换红外光谱仪(FTIR,德国布鲁克公司);Nex ION 300D 型电感耦合等离子发射光谱仪(ICP,美国PerkinElmer公司);TGA/SDTA851e型热重分析仪(TGA,瑞士梅特勒公司,10 ℃/min,空气气氛);ESCALAB 250Xi型X射线光电子能谱仪(XPS,英国ThermoFisher公司);Dataphysics OCA25型接触角仪(德国数据物理仪器有限公司).
1.2 实验过程
1.2.1 改性MOF材料的制备 2-氟基对苯二甲酸(H2BDC-F)参照Huang等[20]报道的方法制备.UiO-66-NH2系列材料参照Biswas等[21]报道的方法制备.MoO(O2)2·2DMF参照Tang等[22]报道的方法合成.
氟改性UiO-66固载钼基过氧化物的制备涉及多个反应步骤,具体合成步骤如Scheme 1所示.
不同氟含量修饰的UiO-66-NH2的制备采用一锅煮的方法.UiO-66-1/4NH2-x/4F的典型制备方法如下:将H2BDC-NH2(0.140 g),H2BDC-F(0.143 g)和H2BDC(0.258 g)按照摩尔比1∶1∶2(总摩尔数为3 mmol)与八水合氯氧化锆(1 g,3 mmol)溶于30 mL DMA中,再加入12 mL乙酸混合均匀,放置在一个聚四氟乙烯高压反应釜中于150 ℃加热24 h.冷却到室温,用DMA和无水乙醇分别离心洗涤3次,收集粉末产物,用无水乙醇进行索式提取,干燥,所得固体产物命名为UiO-66-1/4NH2-1/4F.类似的,在H2BDC-NH2,H2BDC-F 和H2BDC 的总摩尔数不变(3 mmol)的前提下,保持H2BDC-NH2的加入量不变(0.140 g,0.75 mmol),调节H2BDC-F的加入量,H2BDC的加入量随之改变.在相同反应条件下,将得到的系列固体产物命名为UiO-66-1/4NH2-x/4F(x=0,1,2,3).
如Scheme 1所示,过氧化钼的修饰采用后合成修饰的方法.

Scheme 1 Schematic diagram of synthetic procedure of UiO-66 modification with amino and fluorine functional groups as well as post-synthetic modification of molybdenum peroxide
参考文献[23]方法制备了水杨酸亚胺改性UiO-66-NH2(UiO-66-1/4NH2-x/4F-sal).将0.5 g UiO-66-1/4NH2-x/4F 充分分散于10 mL 三氯甲烷中,再加入0.5 mL 水杨醛,于40 ℃条件下磁力搅拌3 d.再将成品用乙腈离心洗涤3~5次,最后在120 ℃条件下真空干燥,得到黄色的UiO-66-1/4NH2-x/4F-sal.
改性UiO-66 后修饰锚定MoO(O2)2的典型步骤如下:称取0.243 g MoO(O2)2·2DMF(0.75 mmol),使其完全溶解于50 mL 无水乙醇中,再加入0.5 g UiO-66-1/4NH2-x/4F-sal,在90 ℃条件下回流搅拌24 h.离心收集固体产物,用无水乙醇洗涤至溶液呈无色透明,在鼓风干燥箱中于80 ℃恒温干燥24 h,得到最终产物UiO-66-1/4NH2-x/4F-sal-Mo.
1.2.2 氧化脱硫反应条件 将一定量的BT,DBT和4,6-DMDBT分别溶于甲苯和正庚烷(体积比2∶3)的混合物中,分别配制出3种硫含量为1000 ppm(1 ppm=1 mg/L)的模拟油品.
在三口烧瓶中加入50 mL 模拟油,再加入一定量的BP 作为内标物(与含硫化合物的摩尔比为1∶1),并加入一定量的催化剂及氧化剂,在一定温度下回流搅拌,定时取样进行气相色谱分析.反应结束后,将催化剂过滤洗涤并于80 ℃烘干.
2 结果与讨论
2.1 催化剂的表征
图1 所示为通过多步改性固载了MoO(O2)2的改性UiO-66 材料的XRD 谱图.其中不同—F 含量及—NH2含量的系列样品的XRD 谱图与UiO-66的单晶X射线衍射数据拟合的粉末XRD谱图[13]的衍射峰位置及相对强度均保持一致,证明改性后的UiO-66材料依旧具备原始的晶型和较高的结晶度,说明多步改性过程后材料仍能保持载体原始结构的完整性.

Fig.1 Simulated XRD pattern from single crystal X-ray diffraction of UiO-66(a),XRD patterns of UiO-66-1/4NH2-sal-Mo(b),UiO-66-1/4NH2-1/4F-sal-Mo(c),UiO-66-1/4NH2-1/2F-sal-Mo(d)and UiO-66-1/4NH2-3/4F-sal-Mo(e)
通过SEM 表征比较了水杨醛改性以及负载MoO(O2)2对样品形貌的影响.如图2(A)所示,采用氯氧化锆作为金属盐合成的UiO-66-1/4NH2为无规则多面体,颗粒粒度较为均一,这与采用四氯化锆作为金属盐制备的氨基改性UiO-66的形貌有一定差异,后者可以得到较为规整的正八面体结构[24].相比之下,UiO-66-1/4NH2-1/4F[图2(B)]的形貌与UiO-66-1/4NH2没有显著差异,颗粒大小较为均一,粒径在50~100 nm 之间,且形态不规整.此外,固载了过氧化钼后的改性UiO-66样品[图2(D)和(E)]的晶粒粒径和形貌均无显著变化,说明多步修饰或氟的引入并未对催化剂形貌和晶粒尺寸产生影响.图2(C)和(F)对比了UiO-66-1/4NH2和UiO-66-1/4NH2-1/4F 材料表面与水的接触角(CA),其中UiO-66-1/4NH2与水的接触角为38.7°,而经过部分氟修饰后的UiO-66-1/4NH2-1/4F材料与水的接触角提高至55.3°,说明氟官能团的改性能够有效的提高材料表面的疏水性能.

Fig.2 SEM images of UiO-66-1/4NH2(A),UiO-66-1/4NH2-1/4F(B),UiO-66-1/4NH2-sal-Mo(D),UiO-66-1/4NH2-1/4F-sal-Mo(E),and contact angles of water on the pressed pellet of UiO-66-1/4NH2(C),UiO-66-1/4NH2-1/4F(F)
不同氟含量修饰前后的UiO-66-1/4NH2系列材料的低温氮气吸附-脱附等温线如图3所示.所有样品均表现为典型的Ⅰ型吸附等温线.表明所制备的材料中有微孔结构,但在较高的相对压力(p/p0>0.8)下,氮气吸附等温线末端产生了滞后环,这是由于纳米尺度的晶粒堆积产生的晶间孔所致.值得注意的是,不同氟含量改性后的系列样品UiO-66-1/4NH2-x/4F(x=1,2,3)的BET 比表面积为1065~1110 m2/g,均高于UiO-66-1/4NH2的比表面积(963 m2/g),说明氟修饰改性并未造成骨架上孔道的堵孔,相反,氟改性提高了材料的BET 比表面积.水杨醛修饰后的系列材料UiO-66-1/4NH2-x/4F-sal 的BET 比表面积下降为770~912 m2/g.

Fig.3 N2 adsorption-desorption isotherms of UiO-66-1/4NH2-x/4F
负载MoO(O2)2之后的催化剂比表面积进一步降低至517~601 m2/g(表1),可以看出,固载金属后材料的BET 比表面积均有明显降低,说明经过水杨醛改性和负载MoO(O2)2堵塞了骨架的部分孔道.ICP 分析结果表明,UiO-66-1/4NH2-sal-Mo,UiO-66-1/4NH2-1/4F-sal-Mo,UiO-66-1/4NH2-1/2F-sal-Mo和UiO-66-1/4NH2-3/4F-sal-Mo中Mo的负载量分别为1,0.8,0.9 和1 mmol/g,说明通过后合成修饰的方法能够较好地控制MoO(O2)2的加入量,氟的引入对UiO-66 骨架疏水性的调控并未对Mo的负载量造成显著影响.
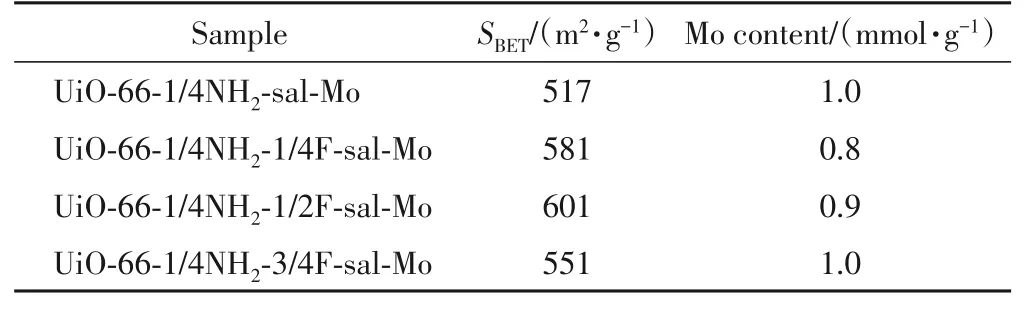
Table 1 BET surface area and ICP analysis results
图4(A)为UiO-66-1/4NH2及不同含量氟官能团改性的系列材料UiO-66-1/4NH2-x/4F 的FTIR 谱图.系列材料的FTIR 谱图中1383,767和579 cm-1处的吸收振动峰可分别归属于—COO、对苯二甲酸配体的芳环上的C—H弯曲振动和Zr—O—C[23].与UiO-66-1/4NH2不同的是,氟官能团改性后的材料在1237和978 cm-1处的吸收振动峰可归属于2-氟对苯二甲酸配体上的C—F 伸缩振动[25].图4(B)对比了固载MoO(O2)2前后样品的FTIR谱图.结果表明,MoO(O2)2引入后,UiO-66-1/4NH2-1/4F-sal-Mo样品中出现了以955 cm-1为中心的吸收振动峰,可归属于Mo=O,表明MoO(O2)2被成功锚定在骨架上[22].采用XPS 图分析了UiO-66-1/4NH2-1/4F-sal-Mo 样品上F 元素的化学状态,结果如图4(C)所示,其中F1s的高分辩XPS拟合谱图的中心峰的结合能为687.0 eV,对应于共价的C—F键[26],这与FTIR光谱表征结果一致.

Fig.4 FTIR spectra of UiO-66-1/4NH2(a),UiO-66-1/4NH2-1/4F(b),UiO-66-1/4NH2-1/2F(c),UiO-66-1/4NH2-3/4F(d)(A),comparison of FTIR spectra of UiO-66-1/4NH2-1/4F before and after modification with MoO(O2)2(B)and XPS deconvolution of F1s of UiO-66-1/4NH2-1/4F(C)
通过热重分析(TGA)研究了UiO-66-1/4NH2和UiO-66-1/4NH2-1/4F在空气气氛下的热稳定性,热失重曲线如图5(A)所示.从图中可见,第一段失重在120 ℃之前,UiO-66-1/4NH2和UiO-66-1/4NH2-1/4F质量损失分别为7.0%和18.2%,此时失重归属于载体孔道内部及表面吸附的乙醇和水分子的脱除.UiO-66-1/4NH2的第二段失重在120~334 ℃区间,质量损失为7.7%,而UiO-66-1/4NH2-1/4F的第二段失重在120~316 ℃区间,这是一段缓慢的失重过程,质量损失仅为3.3%;根据文献[27]报道,第二段失重对应的是部分配体分子的失去,而这部分失重并未对骨架造成不良影响,因此,UiO-66-1/4NH2和UiO-66-1/4NH2-1/4F在空气气氛下分别在334及316 ℃之前结构依旧稳定.继续加热,材料骨架开始发生坍塌,并出现较大失重,分别在530和560 ℃达到完全失重,残余量分别为40.4%和36.2%,此时残余物质应为ZrO2.扣除溶剂分子后,计算得出ZrO2实际占比分别为43.5%及44.3%;而按理想分子式计算其理论残余量应为43.0%和42.4%,可见2种骨架中实际Zr含量均高于理论残余量,即配体占比低于理论值,说明在形成三维多孔晶体骨架的过程中存在着配体缺失即缺陷位点,而配体的缺失会增加Zr的配位不饱和中心从而增强骨架的Lewis 酸性.因此,氟的引入增加了UiO-66骨架的缺陷位,从而增加了UiO-66 骨架的Lewis 酸性,这对氧化脱硫反应起到了促进作用.图5(B)为UiO-66-1/4NH2-sal-Mo和UiO-66-1/4NH2-1/4F-sal-Mo 的XPS 拟合谱图,UiO-66-1/4NH2-sal-Mo 的XPS 拟合谱图[图5(B)谱线a]中,分别在232.6和235.8 eV 处观察到2个峰,可分别归属于Mo3d3/2与Mo3d5/2,其峰面积比约为2/3,说明UiO-66-1/4NH2-sal-Mo 中Mo 以+6 价的形式存在;而在UiO-66-1/4NH2-1/4F-sal-Mo 的XPS 拟合谱图[图5(B)谱线b]中,Mo3d3/2与Mo3d5/2的结合能分别为232.2 和235.4 eV,Mo3d3/2与Mo3d5/2的面积比也为2/3,说明UiO-66-1/4NH2-1/4F-sal-Mo中Mo同样以+6价的形式存在[12].XPS结果表明,氟的引入未改变钼基过氧化物的氧化价态,均表现为+6价;然而氟的引入会导致Mo3d峰向低结合能处偏移,偏移量为-0.4 eV,表明氟改性后有利于电子从载体转移到MoO(O2)2,从而促进催化反应的进行[28].
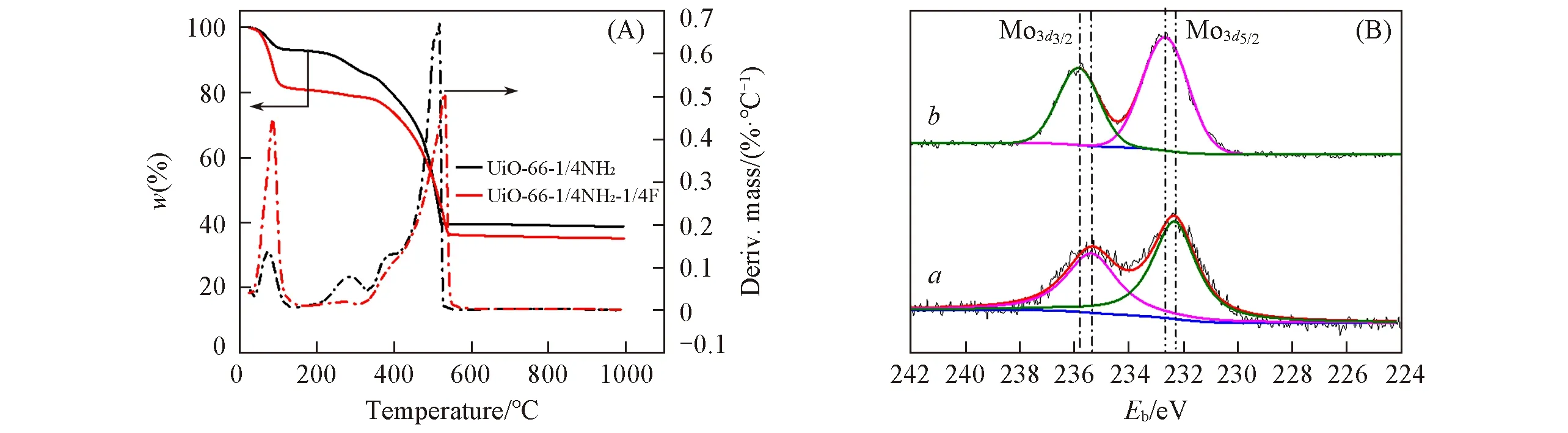
Fig.5 TG-DTG curves of samples in air flow(A) and XPS deconvolution of Mo3d of UiO-66-1/4NH2-sal-Mo(a)and UiO-66-1/4NH2-1/4F-sal-Mo(b)(B)
2.2 催化氧化脱硫
2.2.1 催化剂的初筛 图6(A)对比了相同反应条件下UiO-66-1/4NH2-sal-Mo及不同含量氟官能团改性的系列材料UiO-66-1/4NH2-x/4F-sal-Mo催化氧化DBT的性能.其中UiO-66-1/4NH2-sal-Mo催化氧化DBT 6 h的转化率为55.5%,不同含量氟官能团改性后的催化剂UiO-66-1/4NH2-1/4F-sal-Mo、UiO-66-1/4NH2-1/2F-sal-Mo和UiO-66-1/4NH2-3/4F-sal-Mo催化氧化DBT 6 h转化率分别为89.5%,66.1%和57.7%,均优于没有氟修饰改性的催化剂,其中UiO-66-1/4NH2-1/4F-sal-Mo的活性最高.

Fig.6 Relationship of time and conversion rate of UiO-66-1/4NH2-x/4F-sal-Mo(A)and UiO-66-1/4NH2-1/4F-sal-Mo catalyst with different oxidants(B)
在优选的UiO-66-1/4NH2-1/4F-sal-Mo基础上,进一步考察了不同氧化剂对UiO-66-1/4NH2-1/4F-sal-Mo催化氧化DBT性能的影响.如图6(B)所示,以CHP为氧化剂时,DBT的转化率为89.5%.以TBHP和H2O2为氧化剂,DBT的转化率分别为44.4%和27.9%.文献[29]报道TBHP 作为氧化剂催化氧化脱硫需要较高的反应温度,而H2O2与模拟油品的甲苯/正庚烷混合溶剂不互溶,因此DBT氧化过程受到了界面间混合效率的限制,随着反应时间的延长DBT的转化趋于不变.上述实验结果表明,在本催化反应体系中CHP为最佳的氧化剂.值得一提的是,文献报道了多例基于UiO-66骨架的催化氧化脱硫反应体系,均采用H2O2作为氧化剂,并表现出较好的催化活性,其原因在于溶剂的差异,通过引入乙腈作为单一溶剂或混合溶剂的组成部分,能够将DBT和H2O2萃取至乙腈相进行反应,从而避免了界面间混合的问题[16,17].
2.2.2 反应条件的优化 对优选的UiO-66-1/4NH2-1/4F-sal-Mo催化剂进行了最优条件考察,其中催化剂用量、O/S摩尔比及反应温度是影响DBT转化率的主要因素,考虑以上因素,采用三因素三水平正交实验法L9(33)确定最佳反应条件,正交实验结果见表2,极差分析结果见表3.
根据极差分析得出最优实验条件:催化剂用量为0.1 g,O/S摩尔比5,反应温度80 ℃.影响DBT转化率的主要因素为氧硫比,次要影响因素是反应温度,而催化剂用量对DBT转化率的影响最小.在最佳反应条件下,DBT的氧化脱硫(ODS)转化率达到100%.
2.2.3 不同底物的脱硫性能考察 在优选的反应条件下,考察了UiO-66-1/4NH2-1/4F-sal-Mo催化氧化不同含硫化合物的转化率与反应时间的关系,结果如图7所示.当以BT,DBT和4,6-DMDBT为反应物时,反应6 h内,UiO-66-1/4NH2-1/4F-sal-Mo催化ODS的最终脱硫率分别为66.7%,100%和94.9%.其中DBT 氧化脱硫性能最佳,反应2 h,转化率已达到96%,继续延长反应时间,DBT 转化率趋于平缓.通常情况下,硫化物的氧化与硫原子的电子密度有关,电子密度越高,硫原子的电子密度越高,硫化物的氧化反应越容易发生.DBT,4,6-DMDBT和BT的硫密度分别为5.758,5.760和5.739[30].BT中硫原子的电子密度低于DBT,因此,理论上BT 的转化率应该低于DBT,实验结果证实了这一点.然而,4,6-DMDBT 的转化率与理论分析的预期相反.这可能是由于空间位阻使得底物的硫原子难以接近活性中心所致[31].在反应的初始阶段,BT和DBT的转化率较高,而4,6-DMDBT的转化速率相对偏低,也是因为空间位阻的原因导致反应物到活性位点存在着扩散限制[32].

Table 2 Orthogonal design and experimental results

Table 3 Range analysis table
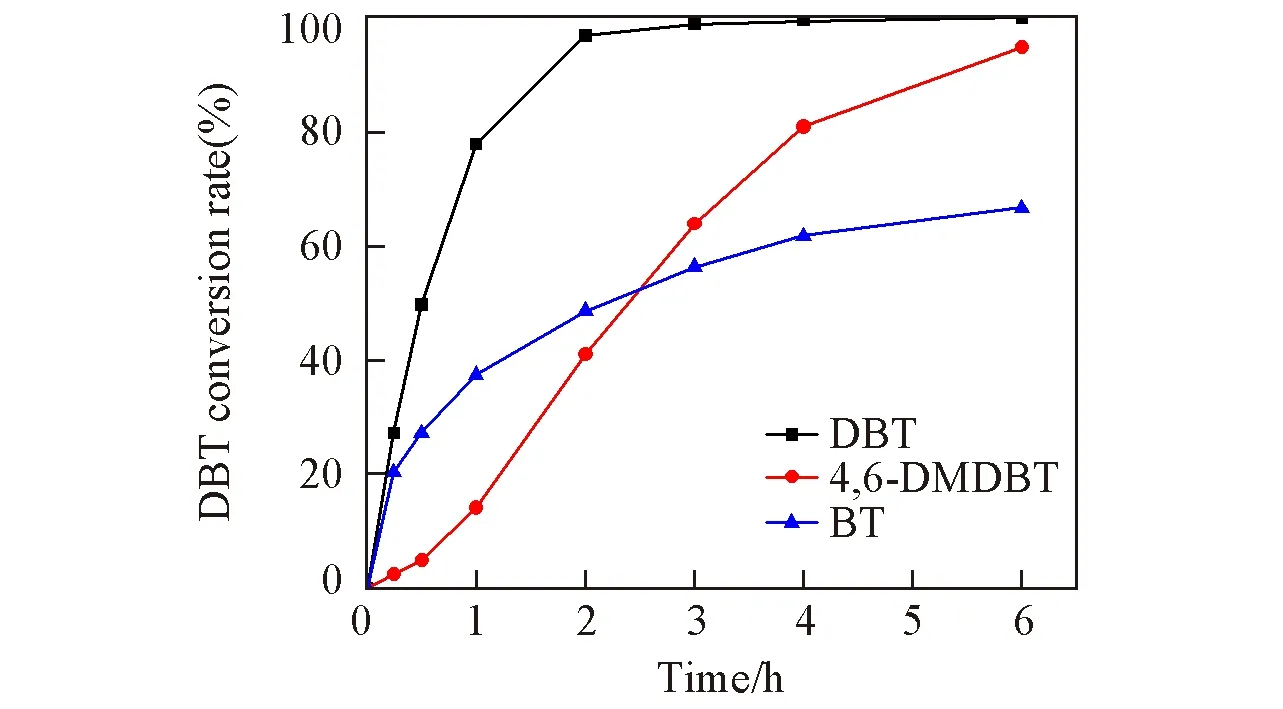
Fig.7 Relationship of time and conversion rate with different reactants

Fig.8 Control experiment of different catalysts
2.2.4 样品的催化活性对比 此外,研究了不同催化剂(UiO-66-1/4NH2-1/4F,UiO-66-1/4NH2-1/4F-sal和UiO-66-1/4NH2-1/4F-sal-Mo)对DBT转化率的影响,结果见图8柱状图a.反应在前述优化的最佳反应条件下进行.在没有催化剂的存在下,DBT不能直接被CHP氧化.值得注意的是,在未引入MoO(O2)2的反应体系中,UiO-66-1/4NH2-1/4F自身具有较高的催化氧化活性,DBT转化率高达85%.其起催化作用的活性中心应为骨架上暴露的Zr4+,热重分析表明,UiO-66-1/4NH2-1/4F骨架上Zr 含量高于理论值,说明存在配位不饱和的Zr4+,由此产生的Lewis 酸性对DBT 氧化过程有显著的催化作用.已有研究表明,通过提高UiO-66材料的Lewis酸性能够提高氧化脱硫的活性.如Ye等[16]通过在UiO-66骨架上引入—NO2官能团,有效地提高了材料整体的Lewis酸性,从而显著提高了氧化脱硫效率.Chakarova等[33]选取了CO,N2和CD3CN 3种探针分子,利用红外光谱研究了UiO-66和UiO-66-NH2的Lewis酸碱性.结果表明,氨基的引入对UiO-66骨架上的酸性影响不大,其原因在于骨架上的Lewis酸性主要来自无机锆氧簇.UiO-66-NH2上的氨基基团没有任何酸性.Xiao等[34]在UiO-66的合成中引入一定量的氨水来制造缺陷位点,通过控制反应时间实现了缺陷中心数量的调控,在DBT氧化反应中,具有一定缺陷位点的UiO-66的DBT转化率明显提高,然而缺陷位增加的同时,催化剂的稳定性下降,使得催化剂的循环稳定性不佳,二次循环的DBT转化率由97%降低至68%.水杨醛修饰后的UiO-66-1/4NH2-1/4F-sal的DBT转化率小于UiO-66-1/4NH2-1/4F 的转化率,这可能是因为水杨基亚胺改性的UiO-66(UiO-66-1/4NH2-1/4F-sal)的空间位阻作用阻碍了骨架上的Zr4+中心与DBT 的接触,从而降低了催化活性.而引入MoO(O2)2后,在MOF骨架上的Lewis酸中心及MoO(O2)2的共同作用下,催化剂UiO-66-1/4NH2-1/4F-sal-Mo效率显著提高,4 h转化率达到100%.
2.2.5 催化剂的循环稳定性 多相催化体系中,催化剂的稳定性是催化剂的关键性能之一.在最优的反应条件下,对UiO-66-1/4NH2-1/4F-sal-Mo 进行了催化剂的循环稳定性考察,单程反应时间为3 h.在每一次反应结束后,将使用过的催化剂离心收集,经二氯甲烷洗涤干燥后再次使用.如图9(A)所示,可以观察到催化剂经过5次循环后催化剂稳定性基本不变,保持在97%左右,说明催化剂具有良好的稳定性能.对比文献中所报道的工作,Zhu 等[35]将UiO-66 用于ODS 反应中,氧化剂为H2O2,实验结果表明,O/S 摩尔比12,催化剂用量为0.1 g和80 ℃的条件下反应150 min,DBT 的转化率接近100%,然而催化剂循环使用5次后,DBT的转化率降低至50%,通过对循环测试后的催化剂进行表征,得出催化剂活性降低的主要原因是砜类化合物在UiO-66上发生强的吸附.而Afzali等[18]报道的MoUiO-66,通过引入醋酸作为助催化剂,能够在室温(25 ℃)下快速实现系列含硫化合物的氧化,然而同样存在着循环稳定性不佳的问题.对比本文的实验及表征结果,氟官能团的引入在增加UiO-66骨架酸性的同时能够有效提高了催化剂的循环稳定性.通过对使用5次后的催化剂进行XRD表征,如图9(B)所示,与新鲜催化剂的XRD谱图相比,循环反应5次后的催化剂的XRD衍射峰位置没有发生变化,说明催化剂骨架结构仍保存完整,然而反应后催化剂的衍射峰强度有所下降,说明多次循环反应后,外层骨架部分破坏造成了催化剂的结晶度下降.通过对反应前后的催化剂(UiO-66-1/4NH2-1/4F-sal-Mo)进行XPS表征,如图9(C)所示,F1s的高分辩XPS拟合光谱的中心峰的结合能为687.0 eV,对应于共价的C—F键,表明催化剂经过多次反应后,其中的氟并未脱落.从图9(D)中可以看出,反应前后催化剂的Mo3dXPS拟合谱图中对应的2个峰中心的结合能位置也未发生改变,说明多次反应后活性中心Mo的价态和配位环境仍然保持稳定.
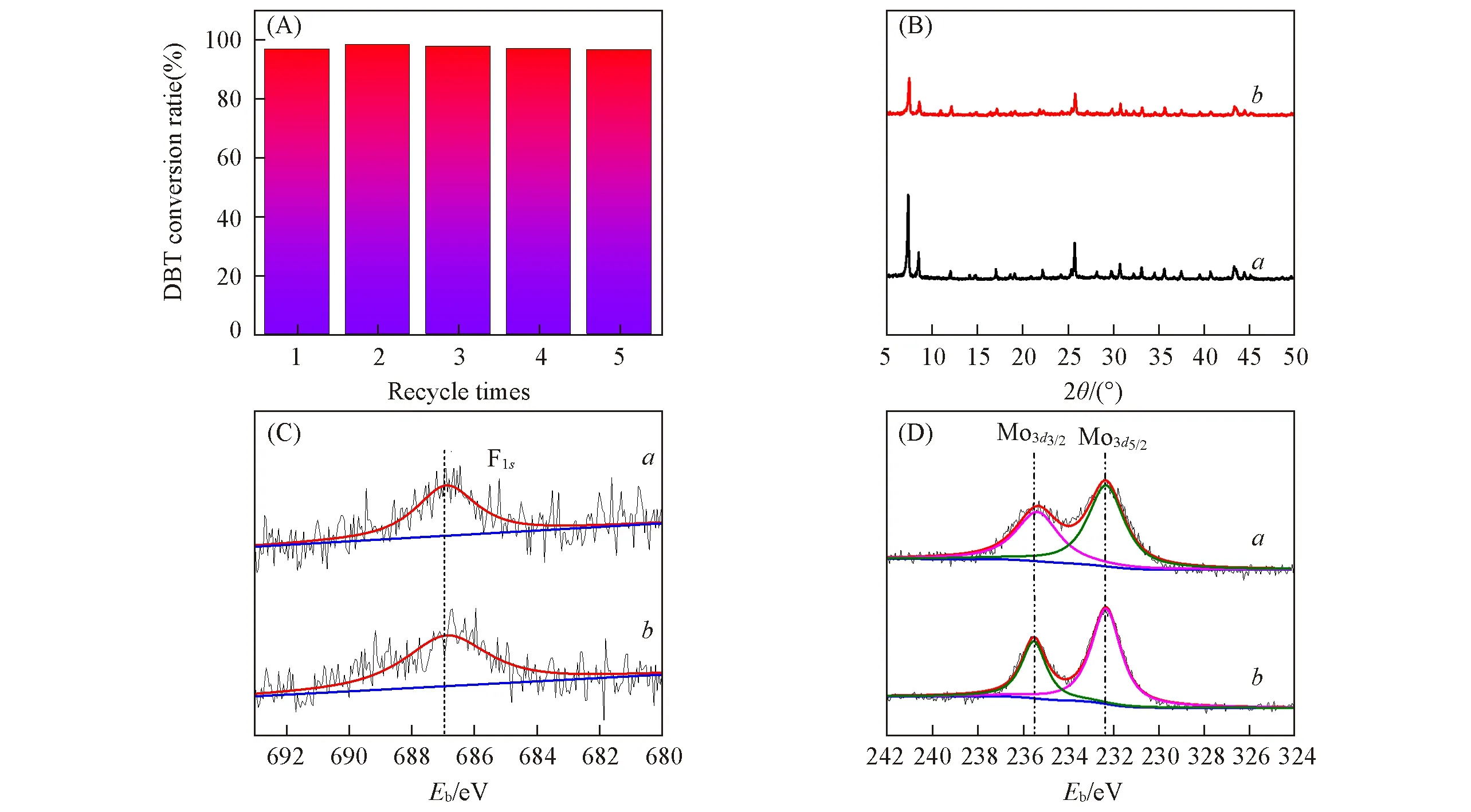
Fig.9 Recycling performance of UiO-66-1/4NH2-1/4F-sal-Mo(A),XRD patterns(B),XPS deconvolution of F1s(C)and Mo3d(D)of fresh(a)and spent(b)catalyst
3 结 论
通过一锅煮的方法制备了双官能团改性的UiO-66,通过多步改性,在UiO-66 骨架上锚定了MoO(O2)2,经过催化剂筛选,其中UiO-66-1/4NH2-1/4F-sal-Mo为最优催化剂.表征结果表明,氟的引入提高了催化剂的疏水性,同时也能增加催化剂的活性位点,达到提高催化剂活性的目的.通过正交实验得到最佳催化氧化脱硫条件为:催化剂用量0.1 g,O/S摩尔比5,反应温度80 ℃,其中氧硫比是影响DBT 转化率的决定性因素.实验与表征结果表明,UiO-66 骨架上含有的Zr 缺陷位点及MoO(O2)2是催化DBT氧化的活性中心.XPS表征结果表明,反应后的催化剂中Mo的价态未发生变化.在优选实验条件下,催化剂经过5次催化循环仍然保持了良好的催化活性及稳定性.
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
电子乐园·上旬刊(2022年5期)2022-04-09
西南石油大学学报(自然科学版)(2021年3期)2021-07-16
环境保护与循环经济(2021年12期)2021-03-16
中国新技术新产品(2020年5期)2020-05-06
中国调味品(2017年2期)2017-03-20
石油知识(2016年2期)2016-02-28
中学化学(2015年2期)2015-06-05
中国煤层气(2014年3期)2014-08-07
化工生产与技术(2014年3期)2014-02-27
