
全谱解卷积核磁共振定量法测定婴幼儿配方乳粉中23种小分子有机物
2021-06-16 10:27王奕寒
食品工业科技 2021年3期
李 玮,王奕寒,姜 洁,林 立
(北京市食品安全监控和风险评估中心,北京 100094)
婴幼儿配方乳粉是以母乳组成的营养元素为标准,以牛乳、羊乳等为原料,适当添加营养素使其组成在数量上、质量和生物学功能上都无限接近于母乳,符合婴儿消化吸收和营养需要的特殊食品。为最大程度地模拟母乳,满足婴幼儿的生长发育需要,生产厂商会按照配方设计添加核苷酸、有机酸、氨基酸等添加剂,这些小分子物质在婴幼儿的免疫调节、提高记忆力、改善肠道菌群及促进机体代谢等方面都发挥重要的作用[1-5]。因此,对配方乳粉中多种小分子物质的测定对乳粉的质量评价和精确模拟母乳成分具有重要意义。
核磁共振定量(Quantitative Nuclear Magneticresonance,qNMR)分析方法以结构测定为基础,是一种可以从整体角度对食品的化学成分进行定性、定量综合分析的现代分析方法。因其非靶向、无偏向的分析特点,qNMR分析方法在食品生产中原料的质量控制、产品稳定性、生产过程中产生的新物质成分及产品研发过程中品质的鉴别等领域,具有很好的检测优势[6-9]。
与目前国标及文献中报道的分光光度法[10-11]、液相[12-14]、气相[15]、质谱[16]等传统检测方法相比,qNMR法能够同时测定多种小分子物质,具有前处理过程简单、取样量小、专属性强等优点。传统的qNMR法是在样品中加入已知量的化合物作为校正参比内标,将被测样品定量质子的信号峰面积与内标物定量信号峰面积进行比较,最终确定样品的浓度[17]。因此,qNMR法的准确性依赖于完整清晰的定量信号峰和准确的定量峰积分面积。然而,婴幼儿配方乳粉的成分复杂,小分子水溶性成分结构相似、含量不高,1H-NMR信号峰会出现信号峰重叠,难以辨认。采用传统qNMR方法进行定量分析时,会遇到无法得到完整信号积分,受相位、基线调节影响大等问题,从而影响定性、定量的准确性。全谱解卷积(Global Spectral Deconvolution,GSD)是一种NMR的信号处理方法,能够在几秒钟内对整个图谱进行解卷积处理,整合复杂峰重叠信号,从而提供完整的峰形、化学位移、耦合常数及积分信息,非常适合1H-NMR信号重叠、信噪比不高的物质定量分析。同时,GSD处理可以降低1H-NMR图谱积分对基线和相位调节的要求,使积分值更加稳定和准确[18]。目前,关于NMR的乳品研究报道多集中在牛乳、人乳的代谢轮廓研究[19-22],少量的婴幼儿配方乳粉NMR定量研究也都采用传统qNMR方法进行定量[23-25]。
本研究首先对婴幼儿配方乳粉水溶液的1H-NMR图谱中的核苷酸、核苷、有机酸、氨基酸等23个小分子水溶性物质进行信号归属,在此基础上,建立了无需对照品的快速、专属、简单的婴幼儿配方乳粉中23个小分子化合物含量的GSD-qNMR同时测定方法。采用建立的定量方法对实际样品进行了测定,以期为今后婴配乳粉的配方研发、产品生产、质量质控、安全检测提供技术支持。
1 材料与方法
1.1 材料与仪器
不同批次厂家的婴幼儿配方乳粉 购于本市超市,均在保质期内,开启后密闭容器内4 ℃保存;重水(D2O,氘代度:99.8%)、3-(三甲基硅基)氘代丙酸钠(TMSP) 美国CIL公司;PBS溶液(pH=7.4) 美国Thermo Fisher公司;超滤离心管(3KD) 美国Millipore公司;Norell 5 mm核磁管 美国Norell公司。
Bruker AVANCE 600 MHZ超导傅立叶变换核磁共振仪(配有CPBBO探头,Topspin 3.2处理软件及60位自动进样器) 美国Bruker公司;XS204电子天平 瑞士Mettler Toledo公司;Centrifuge 5424R离心机 德国Eppendorf公司。
1.2 实验方法
1.2.1 样品溶液的制备 称取2.00 g奶粉样品于10 mL带盖离心管中,加入纯净水5 mL,涡旋3 min,超声15 min后,再涡旋1 min。取涡旋后的液体3 mL于3 kDa的超滤离心管中,9500 r/min转速下离心20 min。取超滤离心得到的样品液体420 μL于5 mm核磁管中,再加入60 μL的重水溶液(含186.8 μg/mL的TMSP)和120 μL的PBS(pH=7.40)溶液,混匀后待测。
1.2.2 仪器条件 定性测定:1H-NMR实验使用Bruker仪器脉冲程序noesypr1d,检测温度25 ℃,设定参数1H的90°脉冲宽度为11.90 μs,谱宽为12019.230 Hz,中心频率为2811.01 Hz,扫描次数为32次。J-分辨、1H-1H COSY、HSQC、HMBC使用Bruker标准脉冲程序,检测温度均为25 ℃,扫描次数分别为 32、16、8、16次。
定量测定:1H-NMR实验使用Bruker仪器脉冲程序noesypr1d,检测温度25 ℃,设定参数1H的90°脉冲宽度为11.90 μs,谱宽为12019.230 Hz,中心频率为2811.01 Hz。脉冲脉冲延迟时间为4 s,扫描次数为96。
1.2.3 样品测定及谱图处理 在1.2.2项实验条件下,调整仪器参数、调谐、控温、匀场、采样及傅里叶变换,得到1H-NMR图谱。测得的1H-NMR谱使用Bruker Topspin 3.2软件处理,变换点数为64 K,LB为1.00 Hz,用指数窗函数处理,基线和相位校正均采用手动方式进行,TMSP为内标信号(δ0.00)。
1.2.4 GSD数据处理 处理后的图谱导入MestReNova(version 12.0,Spain)软件,选择积分校正方法为信号峰校正(Peaks),设置细化等级为等级1,对23个定量物质和定量内标TMSP的定量峰进行解卷积处理,获取各定量峰的峰面积。
1.2.5 定量计算公式 以TMSP为定量内标,分别采用传统的积分方法和解卷积处理后的图谱积分方法按以下公式计算样品中各物质的含量。
式(1)
式中:W为各物质的含量;A为定量峰积分面积;N为定量峰所包含的质子数;M为物质相对分子质量;下标u及t分别为分析物和内标物。
1.2.6 方法学考察
1.2.6.1 标准曲线 称取适量23种标准品置于烧杯中,加水溶解后转移到10 mL容量瓶中,加水至刻度线后摇匀,配制成混标储备液。混标溶液中各物质浓度分别为:缬氨酸1.18 mg/mL、乳酸1.75 mg/mL、丙氨酸1.12 mg/mL、乙酸47.5 mg/mL、柠檬酸35.72 mg/mL、肌酸3.07 mg/mL、肌酐0.83 mg/mL、胆碱6.51 mg/mL、L-左旋肉碱0.85 mg/mL、胞嘧啶核苷0.69 mg/mL、腺嘌呤核苷0.73 mg/mL、次黄嘌呤核苷0.55 mg/mL、乳清酸2.17 mg/mL、马尿酸1.96 mg/mL、尿嘧啶核苷0.31 mg/mL、鸟嘌呤核苷0.66 mg/mL、胞嘧啶核苷酸2.67 mg/mL、尿嘧啶核苷酸2.34 mg/mL、鸟嘌呤核苷酸3.26 mg/mL、甲酸0.13 mg/mL、次黄嘌呤核苷酸0.73 mg/mL、腺嘌呤核苷酸1.79 mg/mL、烟酸0.89 mg/mL。吸取100 μL的混标储备液,加纯净水900 μL后混匀,取500 μL稀释后储备液,在再加500 μL纯净水,依次对倍稀释为5个浓度的混标溶液。取混标溶液420 μL于5 mm核磁管中,再加入60 μL的重水溶液(含186.8 μg/mL的TMSP)和120 μL的PBS(pH=7.40)溶液,混匀。以1.2.2节中的测定条件进行1H-NMR图谱,以积分得到的标准品定量峰面积为Y轴,以加入标准品质量为X轴做线性回归。
1.2.6.2 精密度 按1.2.1节方法制备供试品一份,将制备好的供试品在1.2.2节下条件平行测定6次,计算样品中23个化合物定量峰积分面积的RSD。
1.2.6.3 重复性 取同一品牌、同一批次的奶粉样品,按1.2.1节下方法平行制备6份供试品溶液,在1.2.2节条件下测定1H-NMR图谱,计算样品中23个化合物定量峰积分面积的RSD。
1.2.6.4 稳定性 取同一供试品溶液,分别在制备后0、2、4、8、12、24 h,在1.2.2节条件下测定1H-NMR图谱,计算各时间点图谱的23个化合物定量峰面积的RSD。
1.2.6.5 回收率 平行称取同一品牌的奶粉样品9份,按照1.2.1项下制备供试品溶液,其中3份作为对照,另外6份各加入100 μL稀释后的混标溶液,其中混标溶液中含缬氨酸59.00 μg/mL、乳酸87.75 μg/mL、丙氨酸56.25 μg/mL、乙酸23.75 μg/mL、柠檬酸1786.25 μg/mL、肌酸153.60 μg/mL、肌酐1.75 μg/mL、胆碱325.75 μg/mL、L-左旋肉碱42.5 μg/mL、胞嘧啶核苷34.5 μg/mL、腺嘌呤核苷36.75 μg/mL、次黄嘌呤核苷27.85 μg/mL、乳清酸108.65 μg/mL、马尿酸98.00 μg/mL、尿嘧啶核苷15.45 μg/mL、鸟嘌呤核苷32.95 μg/mL、胞嘧啶核苷酸113.65 μg/mL、尿嘧啶核苷酸116.95 μg/mL、鸟嘌呤核苷酸163.25 μg/mL、甲酸6.75 μg/mL、次黄嘌呤核苷酸36.75 μg/mL、腺嘌呤核苷酸89.85 μg/mL、烟酸44.65 μg/mL。在1.2.2项下条件测定1H-NMR图谱,计算各化合物的加标回收率。
1.3 数据处理
样品检测结果以平均值±标准偏差表示。
2 结果与分析
2.1 信号峰定性归属结果
婴幼儿配方乳粉水提物的1H-NMR图谱见图1A。根据图谱中质子信号的化学位移、耦合常数等信号特征,结合2D-NMR数据及相关文献报道[19-20,25]等信息,共对1H-NMR图谱中23个小分子水溶性成分进行信号归属,它们是缬氨酸、乳酸、丙氨酸、柠檬酸、肌酸、肌酐、胆碱、L-左旋肉碱、胞嘧啶核苷、腺嘌呤核苷、次黄嘌呤核苷、乳清酸、马尿酸、尿嘧啶核苷、鸟嘌呤核苷、胞嘧啶核苷酸、尿嘧啶核苷酸、鸟嘌呤核苷酸、甲酸、次黄嘌呤核苷酸、腺嘌呤核苷酸、烟酸,数据归属详见表1,其中表中加粗数据为用于定量计算的定量峰化学位移。

图1 婴幼儿配方乳粉水提物1H-NMR图谱(A); 解卷积信号提取后得到的23种 小分子化合物的定量峰图谱(B)Fig.1 1H-NMR spectra of infant formulas(A); Quantitative peaks selected by global spectral deconvolution(GSD)in1H-NMR spectrum of 23 low-molecular-weight organic components content注:1.缬氨酸;2.乳酸;3.乙酸;4.丙氨酸;5.柠檬酸;6.肌酸;7.肌酐;8.胆碱;9.L-左旋肉碱;10.胞嘧啶核苷;11.腺嘌呤核苷;12.次黄嘌呤核苷;13.乳清酸;14.马尿酸;15. 尿嘧啶核苷;16.鸟嘌呤核苷;17.胞嘧啶核苷酸;18.尿嘧啶核苷酸;19.鸟嘌呤核苷酸;20.甲酸;21.次黄嘌呤核苷酸;22.腺嘌呤核苷酸;23.烟酸。

表1 婴幼儿配方乳粉中23个小分子有机 成分1H-NMR图谱信号归属表Table 1 1H-NMR data for 23 low-molecular-weight organic components in infant formulas extracts
2.2 GSD提取定量峰
采用1.2.4中GSD数据处理方法提取出婴幼儿配方乳粉水提物中缬氨酸、乳酸、丙氨酸、柠檬酸、肌酸、肌酐、胆碱、L-左旋肉碱、胞嘧啶核苷、腺嘌呤核苷、次黄嘌呤核苷、乳清酸、马尿酸、尿嘧啶核苷、鸟嘌呤核苷、胞嘧啶核苷酸、尿嘧啶核苷酸、鸟嘌呤核苷酸、甲酸、次黄嘌呤核苷酸、腺嘌呤核苷酸、烟酸,23种小分子化合物的定量特征信号,见图1(B),具体化学位移见表1中加黑数据。以内标化合物TMSP为定量内标物,计算23种化合物的含量。
2.3 方法学验证
采得图谱经过傅里叶变换、定标、相位和基线调整后,分别采用GSD法和传统积分两种方式对内标和化合物的定量峰进行积分,按照1.2.5公式计算结果。分别按照1.2.6中方法学考察内容进行方法学考察实验,两种积分方式的方法学验证结果见表2、表3。
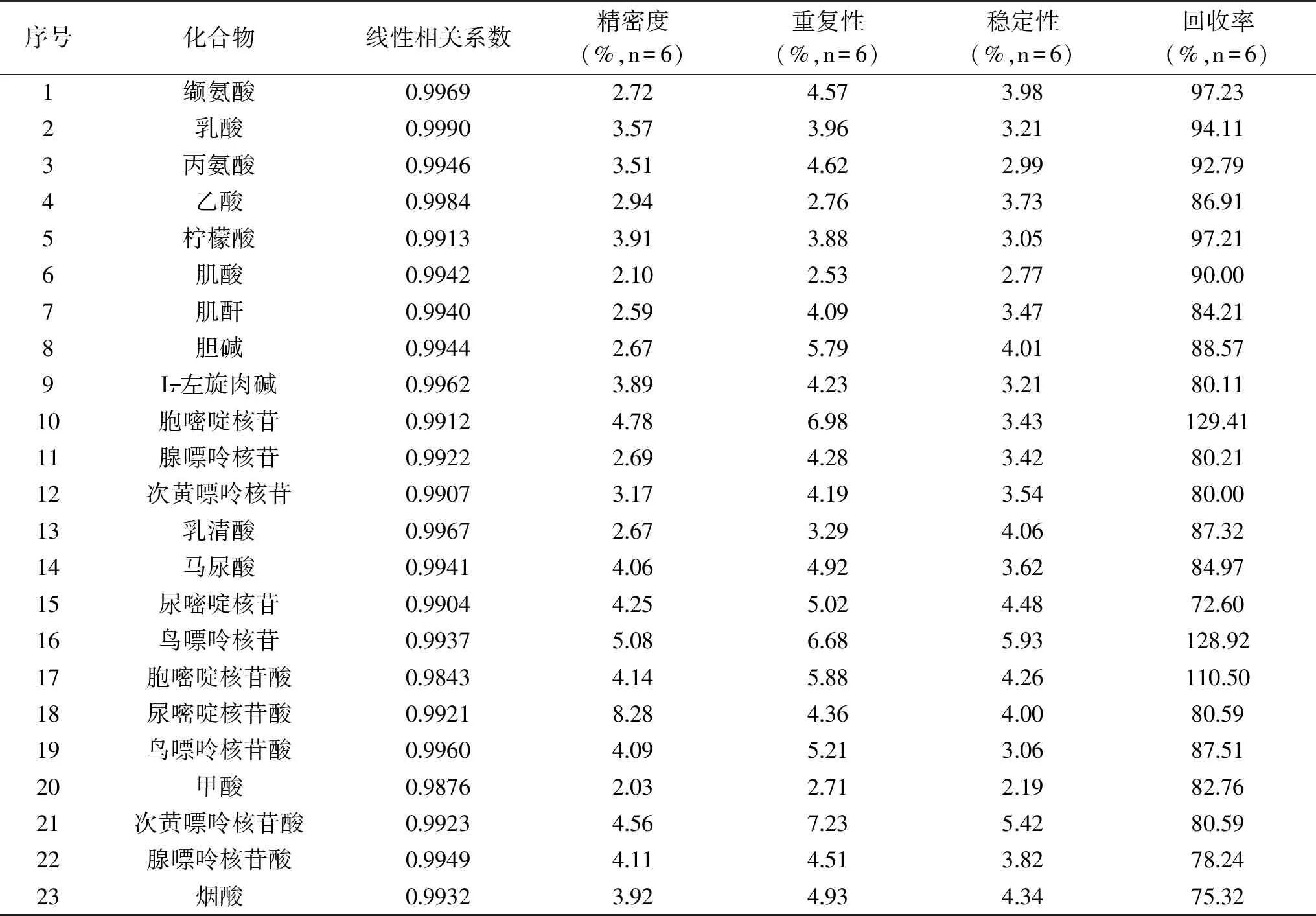
表2 采用传统积分方法处理图谱后的23种化合物定量方法考察学结果Table 2 The results of methodological study for 23 compounds after dealing with the atlas by the traditional integral method
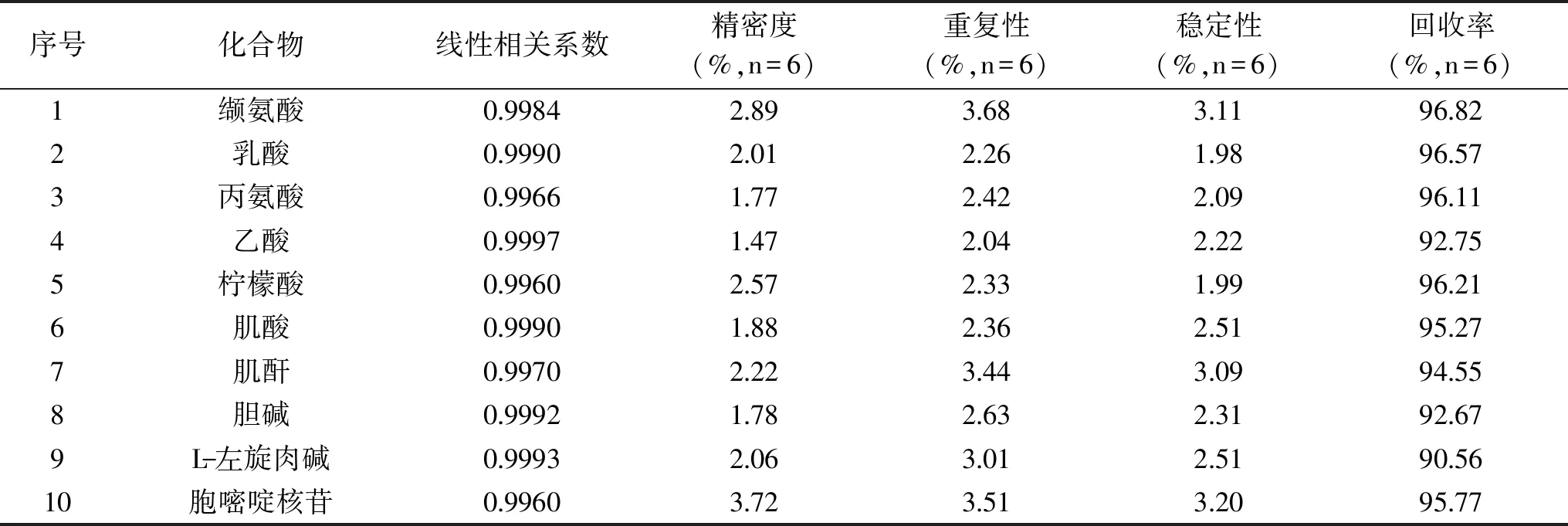
表3 采用GSD方法处理图谱后的23种化合物定量方法学考察结果Table 3 The results of methodological study for 23 compounds after dealing with the atlas by the GSD method

续表
结果显示,采用传统积分方式定量方法的相关系数在0.9843~0.9990之间,精密度的RSD在2.03%~8.28%之间,重复性的RSD在2.53%~7.23%之间,稳定性的RSD在2.19%~5.93%之间,回收率在72.60%~129.41%之间;采用GSD积分方式定量方法的相关系数在0.9960~0.9997之间,精密度的RSD在1.02%~3.72%之间,重复性的RSD在1.34%~3.68%之间,稳定性的RSD在1.62%~3.20%之间,回收率在85.02%~110.00%之间。由此可见,GSD定量方法的精密度、稳定性、重现性和回收率都要优于传统的积分方法,因此,基于GSD处理的qNMR法更适用于婴幼儿配方乳粉中小分子水溶性物质的全面检测。特别是采用了这种积分方式可以减少定量对基线调整的依赖,当内标定量峰与化合物定量峰化学位移相差较大时,效果更为明显。
2.4 样品检测
精密称取6个品牌的乳粉样品,按照1.2.1项下制备供试品,按照1.2.2项下条件测定样品的1H-NMR图谱,采用GSD方法对定量峰进行积分,按照式(1)计算各定量物质的含量,结果见表4。结果显示两种检测方法的测定数值相对标准偏差为3.32%~12.42%,结果较为一致。

表4 6个品牌乳粉样品中23种小分子化合物的含量(mg/100 g,n=3)Table 4 Determination of 23 low-molecular-weight organic components content in infant formulas extracts from 6 manufacturers
3 结论
本研究采用GSD结合qNMR技术建立了婴幼儿配方乳粉水提物中23个小分子水溶性物质的含量测定方法,解决了传统qNMR方法受基线调整及内标峰化学位移影响较大的问题。方法学验证结果表明,23种化合物的线性线性相关系数在0.9960~0.9997之间,平均加样回收率为85.02%~110.00%,且精密度、稳定性和重复性的RSD值均小于4%,均优于传统qNMR方法。在对实际样品的检测中,胆碱测定含量与国标法测定数值相对标准偏差为3.32%~12.42%,较为一致,进一步说明了本方法的可靠性。因此,GSD-qNMR法具有准确、稳定、重复性好优点,适用于复杂食品基质中小分子物质的定量检测。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
肝博士(2022年3期)2022-06-30
食品安全导刊(2021年21期)2021-08-30
昆明医科大学学报(2021年2期)2021-03-29
中国乳业(2020年12期)2020-04-12
中国乳品工业(2019年1期)2019-04-01
中国饲料(2019年19期)2019-03-25
中成药(2017年8期)2017-11-22
中成药(2017年5期)2017-06-13
