
1例先天性佩利措伊斯-梅茨巴赫病临床及基因变异分析
2021-06-11 02:01王金光尚利宏
河南医学研究 2021年13期
王金光,尚利宏
(郑州大学第三附属医院 新生儿科,河南 郑州 450052)
佩利措伊斯-梅茨巴赫病(Pelizaeus-Merzbacher disease,PMD)是一组脑白质营养不良性疾病,其特征是神经髓鞘不能正常形成。PMD是由蛋白脂蛋白1(proteolipid protein l,PLP1)基因不同类型的突变引起的,该基因编码一种主要的髓鞘膜脂蛋白,是一种罕见的弥漫性脑白质髓鞘形成障碍的X性连锁隐性遗传病,1885年首次被报道,男性活婴的发病率为1.45/100 000~1.9/100 000,按临床表现、发病年龄分为6型,先天型、经典型、中间型、无PLP1综合征、复杂型痉挛性截瘫、单纯型痉挛性截瘫[1-2],本文回顾性分析1例先天性PMD患儿的相关外显子检查结果及临床资料。
1 临床资料
患儿,男,5 d,以“出生后发现肌张力低下5 d”为代主诉入院。患儿系第1胎第1产,足月顺产娩出,羊水无污染,出生体质量3 000 g,1分钟Apgar评分为3分,5分钟Apgar评分为8分,在当地县医院诊断为“新生儿肺炎、新生儿缺氧缺血性脑病、新生儿窒息”,治疗5 d后,因四肢肌张力低下无好转,遂至郑州大学第三附属医院,患儿母亲孕期保健未见异常,体健,否认家族遗传性疾病史。入院查体:体温38.6 ℃,脉搏170次·min-1,呼吸 39次·min-1,体质量2 900 g,反应差,营养一般,哭声低弱;前囟1.0 cm×1.0 cm,平软,对光反射灵敏,眼球震颤,口唇红润,两肺听诊双肺呼吸音粗,未闻及明显干湿性啰音,心脏听诊心前区未闻及杂音;腹部平软,肝脾肋下未触及,不能竖头,四肢松软,肌张力低。血生化指标:C反应蛋白明显升高,血氨、同型半胱氨酸、血糖监测未见异常,血气分析无异常,脑脊液未见异常,甲状腺功能无异常。CT示脑白质低密度影,眼底筛查未见明显异常,动态脑电图未见明显异常,心脏彩超示卵圆孔未闭。血液、尿液遗传代谢病筛查未见异常,肝胆及泌尿系超声未见明显异常,头颅MRI提示脑白质髓鞘化障碍(图1),肌电图示部分肌肉失神经改变,脑干听觉诱发电位未通过,视觉诱发电位未通过。结合患儿临床表现及头颅MRI高度怀疑PMD,经患儿家属同意后,抽取该患儿及其父母静脉血各3 mL,送北京贝瑞和康医学检验基因公司进行全外显子组高通量测序,利用贝瑞基因自主研发的Verita Trekker®变异位点检测系统和Enliven®变异位点注释解读系统对数据进行分析,检测到基因PLP1的1个变异,即exon2:c.83G>A:p.G28E(图2),国内外尚未见报道。一代测序验证结果显示:先证者在基因PLP1上发生c.83G>A,半合子变异,母亲发生杂合变异,父亲未发生变异,变异来自母亲,符合X性连锁隐性遗传病孟德尔遗传规律。结合患儿情况及临床表现,确诊为先天性PMD,住院期间给予其对症治疗,患儿体温好转,住院12 d出院(因经口吃奶差带胃管出院),随访至今,目前月龄18个月,不能独坐,四肢松软,无认生表现,语言运动发育明显落后。
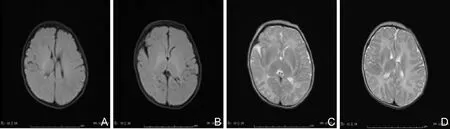
A、B为脑白质在T1W1呈低信号;C、D为脑白质在T2W1呈高信号。
图1患儿头颅MRI

A为患儿PLP1基因测序图,c.83G>A半合子变异;B为患儿父亲PLP1基因测序图,无变异;C为患儿母亲PLP1基因测序图,c.83G>A杂合变异。
2 讨论
PMD是一种X性连锁隐性遗传性脑白质营养不良疾病,男性多发,1885年被Pelizaeus首先报道,1910年德国病理学家Merzbacher对这个家系进行研究,发现该病有X性连锁隐性遗传特点,在1例患者脑组织尸检中发现脑白质髓鞘缺失,故将该病命名为PMD[1]。该病是由于PLP1基因变异导致神经髓鞘合成障碍,表现为髓鞘发育不良,重者缺失,多发生于新生儿[3-4]。运动功能障碍、肌张力低下、眼球震颤、共济失调、发育迟缓、认知障碍等是其常见临床表现,头颅MRI表现为髓鞘化延迟,导致的病理改变是不能形成正常的髓鞘化[1,2,5]。PMD遗传异质性在临床异质性上有较好的反映,该疾病与PLP1外显子和内含子缺失、拷贝数异常及点变异有关。致病基因PLP1位于Xq22.2,长约17 kb,由6个内含子和7个外显子组成。该基因编码PLP1蛋白及其DM20剪切异构体,PLP1蛋白含有276个氨基酸,脑白质髓鞘主要成分是PLP1蛋白,占比约50%,PLP1主要在少突胶质细胞中表达,保障前体细胞发育为脑白质髓鞘。PLP1基因缺陷可导致髓鞘和少突胶质细胞功能异常,继而导致髓鞘形成障碍或少突胶质细胞的死亡,从而导致白质区域髓鞘的减少或缺失。目前,基因组库统计的PLP1基因变异有200余种,包括重复变异、点变异与缺失变异等[1-6]。
有研究表明,PLP1相关疾病谱系中表型与基因型具有明显的相关性,PLP1基因变异包括重复变异、点变异和缺失等。其中重复变异占比最高,占50%~70%,点变异占10%~25%,缺失约占2%,在经典型PMD中重复变异多见,在基因缺失患者中临床表型一般不显著,PLP1基因变异导致神经功能和正常髓鞘化障碍,点突变和拷贝数异常在内的大多数变异都会导致严重疾病。有研究表明,使用CRISPR-Cas9抑制严重PMD点突变小鼠模型中PLP1表达,可促进髓鞘形成,并将小鼠的神经传导速度、运动功能和寿命恢复至野生型水平,PLP1抑制可以发展为治疗人类PMD的方法,基于寡核苷酸的治疗剂可以在体内传递至少突胶质细胞以调节神经功能和寿命,为髓磷脂疾病提供了新的药物治疗方式[7]。PMD头颅MRI具有典型性改变,表现为髓鞘化异常,髓鞘化形成延迟,远远落后于同胎龄儿[8-9],在脑白质T1W1、T2W1上常常表现为髓鞘化障碍,该影像学检查对PMD的诊断有很大价值。
此外,佩梅样病(Pelizaeus-Merzbacher-like disease,PMLD)与PMD临床表现相似,是一种常染色体隐性遗传病,在男女中发病率无差异,致病基因为GJC2基因,该基因编码CN47,该蛋白在中枢神经系统髓鞘化形成中起重要作用。由于这两种疾病临床特征和头颅MRI表现类似,难以区分,最优的鉴别方式为基因检测[10]。
截至目前,二代测序全基因组外显子检查能够很好地检测各种点变异[11],经查询现有HGMD、Clinvar数据库,该患儿携带的PLP1上发生c.83G>A半合子变异未见相关文献报道,为非同义变异,变异位点致病性评级及数据解读规则参考美国医学遗传学与基因组学学会指南,排除千人基因组、ExAC、gnomAD、贝瑞基因中国人群特有数据库(神州基因组数据库)等数据库中变异频率大于1%的变异位点,去除非功能性变异位点(同义变异、非编码区变异等),再经过致病性预测(SIFT、PolyPhen-2、CADD等软件)、临床症状对照、相关疾病数据库查询与文献参考等综合考虑,找到候选基因变异位点进行家系验证,该变异发生在PLP的conserved site功能结构域,该变异在贝瑞基因中国人群特有数据库(神州基因组数据库)、ExAC、千人基因组和gnomAD中未被发现,该变异与已确定为致病性的变异发生在同一位置,但有不同的氨基酸错义变异,经GERP等对其进行保守性预测,结果显示该位点进化上保守,具有潜在的功能影响,经SIFT和PolyPhen等对其进行蛋白功能预测,结果显示为有害。但相同氨基酸位点的其他形式变异(c.83G>T:p.G28V)是HGMD数据库收录的与PMD相关的变异,结合患儿临床表现及头颅MRI结果,诊断先天性PMD。
综上所述,该患儿生后即出现四肢肌张力低下,眼球明显震颤,运动发育落后,吞咽功能障碍,头颅MRI示脑白质未见髓鞘化,基因检测得以明确诊断,目前PMD尚无有效治疗方法,康复治疗能改善患儿生活质量,有动物模型研究表明基因方法治疗能改善预后,相信不久的将来基因治疗能应用于PMD患儿,本研究通过分析该患儿的临床资料及其父母的PLP1基因明确诊断为先天性PMD,为该家庭提供遗传咨询,对优生优育提供帮助,该研究发现1个新变异位点,丰富了PLP1基因变异谱。
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24
磁共振成像(2022年8期)2022-10-08
中西医结合心脑血管病杂志(2022年14期)2022-08-19
健康大视野(2020年7期)2020-04-26
医学新知(2019年4期)2020-01-02
保健与生活(2019年15期)2019-09-12
党的生活(黑龙江)(2018年9期)2018-10-17
东坡赤壁诗词(2018年3期)2018-07-16
健康大视野(2018年22期)2018-02-18
扬子江(2016年1期)2016-05-19
