
外力对三元哈斯勒半金属ZrIrSb电子结构和光学性质影响的第一性原理研究*
2021-06-01 03:32韦俊红孔慧君张敏平
河南工学院学报 2021年1期
韦俊红,孔慧君,刘 潇,张敏平
(1.河南工学院 理学部,河南 新乡 453003;2.河南师范大学 物理学院,河南 新乡 453007)
0 引言
能源问题一直是全世界关注的问题,再生能源、环保能源和新型能源的开发和研究随着全球经济的发展以及能源需求量的逐日增加而变得极其重要。近年来,有机-无机钙钛矿太阳能电池材料引起了人们的关注,尤其在新兴的光伏技术领域,它提供了高效率的固态太阳能电池材料[1-5]。有机钙钛矿MAPbI3(MA=CH3NH3+)与混合的卤族化合物MAPb(I1-xBrx)3、MAPb(I1-xClx)3在电池领域具有广阔的应用前景[6-8],但是材料中的Pb原子具有毒性,对环境有污染,因此探寻经济环保、可再生、便利高效的无机太阳能吸收材料是新能源研究领域的一个艰巨任务。
近年来,作为一种新颖的能源材料,三元哈斯勒半金属材料引起了大家的关注。由于其在高温下具有较高的机械稳定性而被认为是潜在的热电材料[9-11]。2015年,Zunger[12]等报道了三元哈斯勒半金属化合物大家族被遗漏的一些化合物,并且发现这些化合物中存在一些有趣的物理性质,比如拓扑、热电、压电、光学等性质。在改变晶格或者原子替代的情况下,HfIr(As, Sb, Bi)[13]已经被预测出会发生拓扑相的转变;通过晶格常数畸变或者用大小不同的原子进行替代来实现加外力,打破晶体的空间反演对称性,导致HfIrBi在费米能级处能带发生劈裂并出现Weyl点,Weyl点的出现使HfIrBi成为Weyl半金属材料;化合物ZrNiPb、HfNiPb、HfPdPb[14]的带隙较小(<0.5 eV),实验测出ZrNiPb在室温下是良好的热电材料,被认为是一种具有潜力的透明导电体;TaIrGe[15]是一种P型透明导体,实验测出该材料在3.36eV左右有很强的光吸收峰,理论上也被证明是好的热电与光学材料。报道中提到,ZrIrSb[12]属于一种透明的P型半导体材料,具有罕见的高空穴电导率,有很好的光吸收效率。对于光学性质,合适的带隙可以有效地提高材料的光吸收效率。而对带隙的调控,除了尺寸的改变,应力已经成为微电子领域中半导体带隙设计的一个常规手段,并且带隙的调控可以从根本上改变材料的电子性质。理论上一系列的研究已经证明了加力可以改变材料的性质,如β-Na0.33V2O5[16]、 β-K2Bi8Se13[17]、Bi2Te3[18]等材料在外力的作用下可以有效地调控带隙,从而提高材料的相关性质。从前期的研究我们可以看出,ZrIrSb是一种非常有潜力的半导体材料,因此,研究应力对ZrIrSb结构以及材料性质的影响就显得非常重要。本文采用密度泛函理论,系统研究ZrIrSb在不同外力下能带结构以及光学性质的改变。
1 理论方法
为了更好地研究ZrIrSb的性质,我们采用了基于密度泛函理论(DFT)[19]的VASP (Vienna ab initio simulation package)[20]软件包对ZrIrSb进行了结构优化。
所有的晶格优化和原子位置弛豫时的交换关联势都采用广义梯度近似(GGA)[21]来处理,最小力设置为10-5Ry/Bohr。由于传统的广义梯度近似或者局域密度近似势(LDA)会低估半导体的带隙,而精确的能带结构对于模拟材料的性质极其重要,为了避免能带被低估,我们采用改进的mBJ (modified Becke-Johnson)[22]交换关联势进行自洽和计算材料的光学性质。ZrIrSb的电子结构能带图和光学性质的计算采用了WIEN2k[23]软件包,截断能设置为7.0 Ry,进而达到能量收敛的目的。球内的价波函数扩展到最大值L=10,电荷密度作为傅立叶级数最多G=12。自洽过程中,第一布里渊区的K点网格设置为18×18×18。
2 结果与讨论
2.1 电子结构
三元哈斯勒半金属化合物ZrIrSb的空间群号是216(F-43m),结构如图1所示。其中,Zr原子和Ir原子处在邻近的四面体中,而位于4b位置的Ir原子和4a位置的Sb原子形成了闪锌矿型晶格结构,Zr原子位于剩下的面心立方4c的位置。Zr、Ir和Sb的原子位置坐标分别为4c(0.5,0.5,0.5),4b(0.25,0.25,0.25)和4a(0,0,0),剩余空位的位置为(0.75,0.75,0.75)。
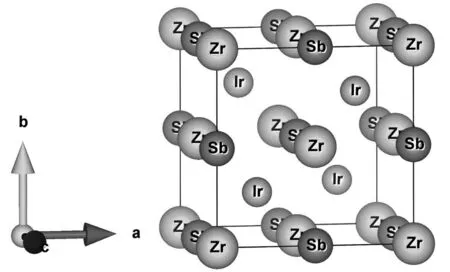
图1 ZrIrSb结构侧视图
ZrIrSb的电子排布为s2p6d10,每个分子式价电子数是18,一般呈现的是半导体或者半金属状态。首先对ZrIrSb的晶格进行优化,结构弛豫后达到能量最低的稳定态时晶格常数为6.359Å,与先前报道的值6.372Å误差范围为0.2%,吻合得较好[24]。基于优化后的晶体结构,我们研究了ZrIrSb沿着布里渊区高对称点W-L-G-X-W-K的能带结构。由图2 的分波投影能带结构可知,ZrIrSb是一种具有间接带隙的半导体材料,带隙为1.52 eV,此结果值与ZrIrSb相关研究的结果非常相似[12,24],能带值略大于采用GGA[24]方法得到的结果,而略小于HSE06[12]得到的结果。HSE06是大家公认的计算能带最准确的方法,但是这种方法存有弊端:计算光学性质时非常耗时。均衡计算结果的精确度和硬件条件,我们选用了mBJ交换关联势计算材料的电子结构、光学性质。图2中,位于费米能级上方的高对称Γ点(标记为A),主要是由Sb原子的s轨道组成,位于价带最高点的Γ点主要由Zr原子的Dt2g轨道构成,而B、C标识位置处主要是由Zr、Ir原子的d轨道组成的杂化态。ZrIrSb的能带结构特点主要有两个方面:一是具有较大的带隙;二是属于间接带隙,而非直接带隙。A点标识处主要由s轨道组成,而s轨道对外力比较敏感,施加外力时,电子结构容易改变。因此,我们在后续的计算中,需要考虑对ZrIrSb施加外力,研究外力对材料电子结构以及性质的影响。
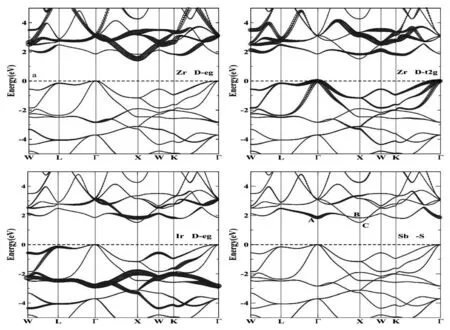
图2 ZrIrSb分波投影能带图
2.2 外力对电子结构的影响
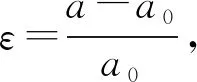
图3给出了ZrIrSb在一系列外力作用下的能带结构。图中表明,在不同外力作用下,ZrIrSb的电子结构发生改变,带隙也随着发生改变。当压力逐渐增大时,间接带隙逐渐增大,带隙Eg从1.54eV增大到1.61 eV。当拉力逐渐增大时,带隙变小,Eg从1.50 eV减小到1.37 eV。当ε=2%时,导带底从高对称点X转变为Γ点,能带结构从间接带隙转变为直接带隙。因此,拉伸应变ε=2%是ZrIrSb带隙转变的临界值。
为了解释ZrIrSb的结构在外力作用下变化的原因,我们重点研究了图2 ZrIrSb的投影能带结构。图中,位于CBM的高对称点X点(C标识处),主要由Ir和Zr原子的d轨道组成,而A标识处由Sb的s轨道组成,s轨道比d轨道对外界的应变更敏感。因此,当对ZrIrSb施加压力时,s轨道与d轨道的能量都会上升,但是s轨道要比d轨道上升得快,在Γ点处由s轨道贡献的能带更容易被提升。当应变从-1%逐渐变化到-5%,s轨道由于能量增加而逐渐升高,d轨道由于能量变化甚微可忽略其在X点的变化。因此,在施加压力时,C标识处能带性质未发生变化,A标识处能量上升,带隙增加。增加拉力时,位于导带A处的s轨道能量下降比C处d轨道能量下降得快,当ε=2%,导带最低点由高对称点C处转变为A处,ZrIrSb的能带结构从间接带隙转变为直接带隙。即随着拉力增加,带隙逐渐减小。一系列的结果表明,应变可以用来调控ZrIrSb的带隙,压缩应变导致带隙增大,拉伸应变导致带隙减小。
2.3 外力对ZrIrSb光学性质的影响
直接带隙的半导体材料,由于价带顶和导带底具有相同的波矢量值,声子未参与能带间的跃迁,致使材料的光学性质活跃,具有很高的光吸收效率[25]。因此,直接带隙的半导体材料由于光吸收效率高而成为太阳能电池的首选材料。原始的ZrIrSb是一种间接带隙材料,对其施加合适的外力就可以把间接带隙转变为直接带隙,其光吸收效率也由此得到提高。因此研究ZrIrSb在各种应变下的光学性质对理解该化合物的光电子性质至关重要。
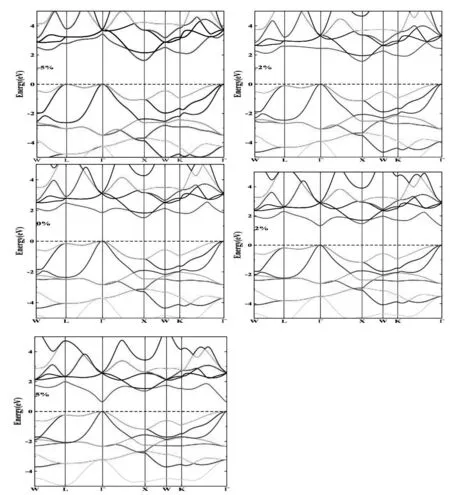
图3 ZrIrSb在不同外力作用下的能带结构图
半导体材料的光学性质可通过复介电函数ε(ω)=ε1(ω)+iε2(ω)计算得到,材料的光学性质与材料的结构紧密相关。ε1(ω)、ε2(ω)是介电函数的实部与虚部。ε1(ω)可以通过Kramer-Kronig关系由ε2(ω)求出来,ε2(ω)通过联合态密度和波函数的所有可能发生电子跃迁的占据态和非占据态间的动量矩阵元计算得出, 计算公式[26]如下:
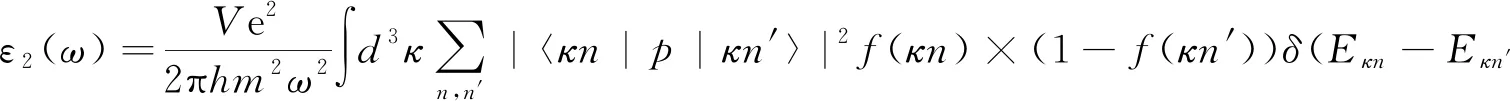
(1)
其中,ћω是光子能量,p是动量算符,|κn〉是晶体波函数,f(κn)是费米函数。
折射率由介电函数可得,公式如下:
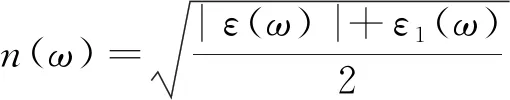
(2)
我们利用Wien2K软件包研究了ZrIrSb在不同外力下的光学性质。光学性质包括复介电函数、折射率和光吸收效率,如图4所示。
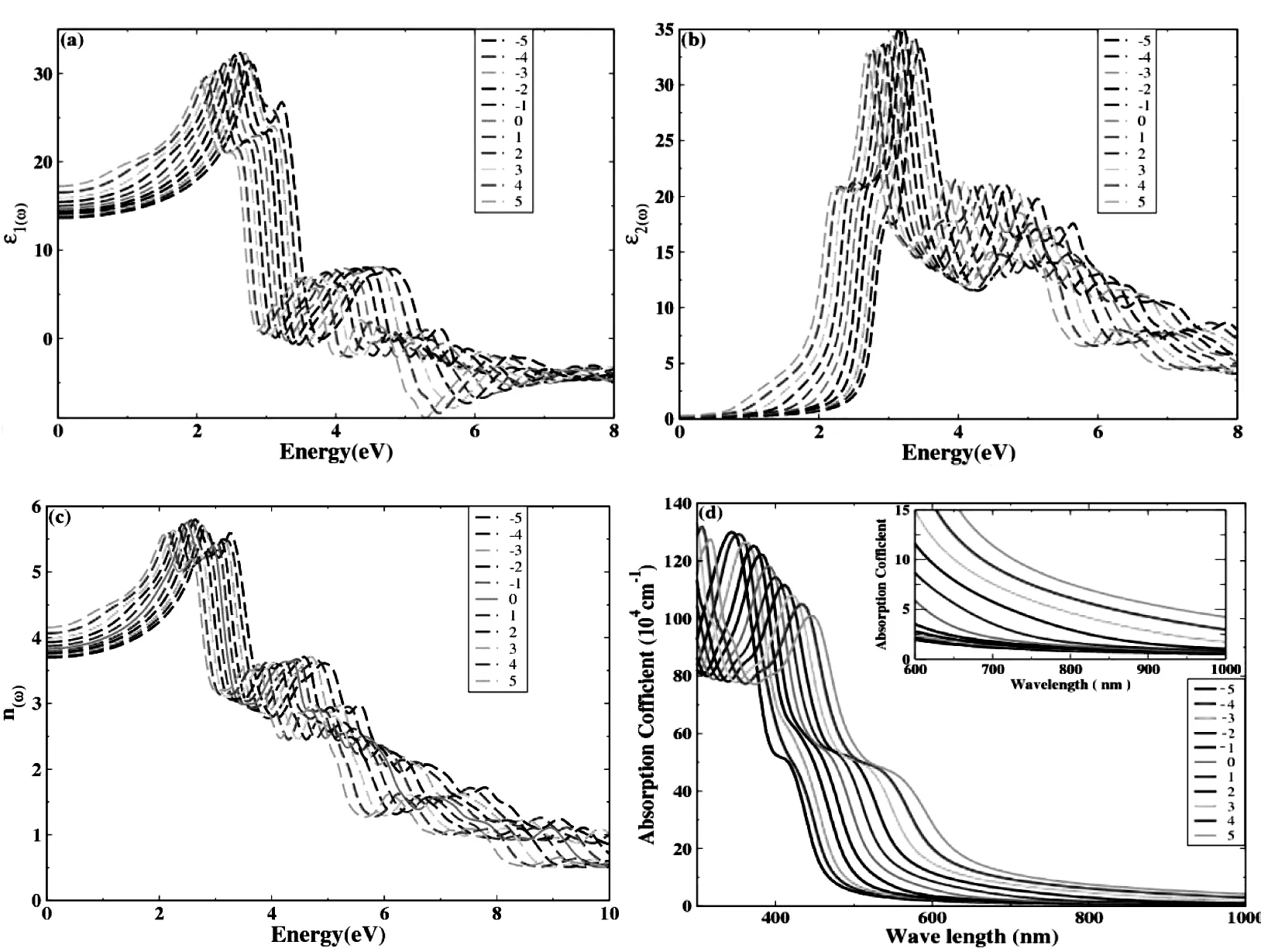
图4 ZrIrSb在不同应变下的光学性质
图4(a)显示的是介电函数的实部ε1(ω)在不同应变下的变化曲线图。在无应变时,ε1(0)的值为14.66。当外力逐渐从压力变为拉力,即ε从-5%到5%的变化过程中,ε1(0)从13.65逐渐增大到17.24,横轴能量先逐渐增加,增大到2.51eV左右达到最大值(ε=-2%),随后逐渐减小。
图4(b)是介电函数虚部ε2(ω)在不同应变下的变化曲线图。从图中可以看出,未加应变时ZrIrSb介电函数阈能(第一临界点) 是2.68 eV。当压缩应变逐渐增大,即ε从0到-5%时,阈值逐渐增加,当ε=-2%时最大;当拉伸应变逐渐增大,即ε从0到5%时,阈值逐渐减小。阈能值对应于能带中Γ与X间的电子跃迁,即价带顶Ir-d和Sb-s与导带底Zr-t2g间的电子跃迁。带隙随着应变的增加而发生变化,阈能值随着带隙的变化而发生移动。
图4(c)是ZrIrSb在不同应变下折射率的变化图。未加应变时,ZrIrSb的折射率的峰值与介电函数实部的峰值位置比较接近, 均位于2.20 eV 附近。在应变的作用下,两者峰值移动的规律基本一致。从曲线图可以看出,无应变时n(0)的值为3.83,当应变逐渐从压缩转向拉伸的过程中,即ε从-5%到5%的过程中,纵轴n(0)的值逐渐从3.62变化到4.15。而横轴能量峰值位置逐渐向左移动,当ε=-2%时,峰值达到最高点,能量值为2.52 eV。
图4(d)为ZrIrSb在不同应变下光吸收效率的变化图。由图可知,应变从-5%到5%变化的过程中,光吸收效率逐渐减小,当应变ε≥2%时,在可见光范围内吸收强度增加,并在吸收光边沿出现红移现象。从两个方面均可以解释红移现象:一是当应变从-5%到5%的变化过程中,带隙随着应变的变化逐渐减小,带隙值在1.62—0.66eV范围内得到了很好的调控,更有利于电子从价带跃迁到导带,吸收光范围增大,从而提高了光吸收效率,导致了红移;从材料的电子结构来看,对于间接带隙,价带导带间的电子跃迁需要声子辅助,而直接带隙则不需要;二是当拉伸应变ε=2%时,ZrIrSb由间接带隙转变为直接带隙,直接带隙光学性质活跃,更有利于电子跃迁,进一步提高了光吸收效率。
3 结论
基于第一性原理,我们计算了三元哈斯勒半金属化合物ZrIrSb在不同应变下的电子结构以及光学性质。结果表明,未加外力时,ZrIrSb是间接带隙的半导体,能带结构与相关实验结果一致。当应变从-5%到5%逐渐变化时,带隙得到了很好的调控,ε=2%时,电子结构由间接带隙转变为直接带隙。结构的变化更有利于电子发生跃迁,随着拉伸应变的增大,光吸收范围增大,吸收效率提高,导致红移现象出现。这一现象表明,ZrIrSb在拉伸应变的作用下有望成为太阳能电池或者光电器件的有用材料。我们的计算结果为后续材料试验提供了可靠的依据。
猜你喜欢
小天使·聪聪画刊(2021年2期)2021-09-10
陕西科技大学学报(2020年6期)2020-11-25
汽车零部件(2020年10期)2020-11-09
人工晶体学报(2020年9期)2020-10-21
无线互联科技(2019年15期)2019-11-07
汉语世界(The World of Chinese)(2019年6期)2019-09-10
科技创新与应用(2018年21期)2018-09-14
汽车生活(2018年5期)2018-06-21
振动工程学报(2017年4期)2018-05-31
电子制作(2018年1期)2018-04-04
