
基于低共熔溶剂的分散液液微萃取/超高效液相色谱-串联质谱法测定马肉中非甾体抗炎药
2021-05-27 03:38郑书展任彩霞张春艳王晓敏盛万里
分析测试学报 2021年5期
郑书展,任彩霞,张春艳,王晓敏,盛万里
(呼和浩特海关技术中心,内蒙古 呼和浩特 010020)
非甾体抗炎药(NSAIDs) 在抗炎、止痛、退热和抗凝血等方面疗效显著,是目前动物养殖业中使用最广泛的兽药种类之一[1]。常见的非甾体抗炎药有苯基丁氮酮、氟尼辛、萘丁美酮、萘普生、托芬那酸、乙酰氨基酚等。近年的研究表明这类抗炎药具有明显的毒副作用,会引起胃肠道、血液、肾脏系统等的不良反应,其中较常见的是肠胃溃疡类严重副作用[2]。NSAIDs可在动物肌肉、牛奶、肝脏和脂肪中积累,会通过食物链对消费者健康构成威胁[3]。其残留问题已引起各国食品安全监管部门的重视,美国、欧盟、日本等国家均对该类抗炎药的最大残留量进行了限定[4]。
马肉肉质鲜嫩,脂肪较少,有独特的鲜香味道,是一类营养价值较高的肉类食品[5]。20世纪60年代以来中国马肉产量逐渐提高,到2010年,中国已成为世界马肉最大生产国。近年来,我国马肉进口量也呈上升趋势,以我国进口蒙古国马肉唯一指定口岸二连浩特为例,2017年进口2.3万吨,2018年进口达2.9万吨[6-7]。然而在马的饲养过程中存在非法使用非甾体抗炎药的情况,2013年欧洲“马肉风波”中,欧盟检测结果显示有0.5%的待测马肉检出含有苯基丁氮酮[8]。而苯基丁氮酮会引起人类再生障碍性贫血,国际上已严禁用于食用动物中。为有效监控马肉品质,有必要建立更为快速、高效的马肉中非甾体抗炎药残留的检测方法。
目前,非甾体抗炎药残留的检测方法有高效液相色谱法、液相色谱-串联质谱法、气相色谱-串联质谱法、毛细管电泳法、薄层色谱法和分光光度法等[9-16]。其中,液相色谱-串联质谱法因灵敏度和选择性高在NSAIDs残留分析中最为常用。非甾体抗炎药的样品前处理方法主要有固相萃取[17-19]、液-液萃取[20]和固相微萃取等[21-22],但这些前处理方式存在操作繁冗、时间长、溶剂用量大、不环保、成本高等不足。分散液液微萃取(Dispersive liquid-liquid microextraction,DLLME)是一种集采样、富集、分离于一体的样品前处理技术,具有萃取溶剂用量小、成本低、富集效果好、操作简便和环境友好等优点,已在残留检测中受到广泛关注[23-24]。各类新型DLLME萃取剂不断涌现[25],其中低共熔溶剂(Deep eutectic solvent,DES)由于其毒性低、可降解、原料丰富、价格低廉、制备简单的特点得到较多应用[26-30],例如,Dil等[31]开发了基于DES的人尿中甲芬那酸含量的测定方法:Shishov等[32]将DES用于牛奶中非甾体抗炎药的测定。然而,现有文献均聚焦于液态样品的前处理,目前尚无基于DES的分散液液微萃取技术用于动物肌肉等固态样品中非甾体抗炎药的检测报道。
本研究以廉价易得的氯化胆碱和苯酚为原料经过简单混合合成疏水性DES,并将其作为萃取溶剂,建立了分散液液微萃取结合超高效液相色谱-串联质谱技术测定马肉中10种非甾体抗炎药的方法。该方法操作简单、准确高效、环保节约,为拓展兽药残留检测的样品前处理技术提供了新的思路。
1 实验部分
1.1 仪器与试剂
1260-6460超高效液相色谱-串联质谱仪(美国安捷伦公司),ST16R冷冻离心机(美国Thermo公司),Vortex-Genie 2涡旋振荡器(美国Scientific Industries)。苯基丁氮酮、氟尼辛、萘丁美酮、萘普生、双氯芬酸、舒林酸、酮洛芬、托芬那酸、吲哚美辛、乙酰氨基酚标准品购自德国Dr.Ehrenstorfer 公司,纯度均大于98%。氯化胆碱(ChCl,分析纯,麦克林公司),苯酚(分析纯,天津风船化学试剂科技公司),抗坏血酸(分析纯,阿拉丁公司),乙腈(色谱纯,安谱公司),实验用水为Milli-Q 超纯水,含10 mmol/L抗坏血酸的20 mmol/L醋酸铵缓冲液(乙酸调至pH 3.5)。
1.2 标准溶液的配制
将各标准品用乙腈稀释成1.0 μg/mL的标准储备液,于冰箱4 ℃避光保存。临用前以乙腈混合逐级稀释成低浓度混合标准工作溶液,使用时以乙腈稀释成所需浓度的标准工作液。

表1 低共熔溶剂的合成Table 1 Synthesis of deep eutectic solvents
1.3 DES的制备
按照表1中的比例分别称取氯化胆碱和氢键供体,置于50 mL具塞试管中,其中DES-1~DES-5采用60 ℃加热搅拌方式,DES-6~DES-9采用室温涡旋振荡混合,直至得到透明澄清的均一液体。
1.4 样品前处理
取代表性马肉样品约500 g,用组织捣碎机充分捣碎混匀,装入洁净容器作为试样,置于-18 ℃冷冻避光保存。称取2.5 g搅碎均匀的试样,加入5 mL含抗坏血酸的醋酸铵缓冲液,混匀振荡10 min,加入1.0 g氯化钠和0.8 mL低共熔溶剂,混匀1 min,再加入0.8 mL分散剂乙腈,涡旋混匀1 min,10 000 r/min离心5 min。取上清液,用少量乙腈定容至1.0 mL,过滤膜后待测定。
1.5 色谱-质谱条件
色谱条件:Acquity UPLC BEH C18(2.1 mm×50 mm,1.7 μm)柱,流动相:A为含5 mmol/L乙酸铵+0.1% 甲酸的水溶液,B为乙腈。梯度洗脱程序:0.0~3.0 min,20%~60%B;3.0~4.0 min,60%~90%B;4.0~4.5 min,90%~20%B;4.5~7.0 min,20%B;流速:0.2 mL/min;进样量:5 μL。
质谱条件:三重四极杆质谱,电喷雾(ESI)电离源,采用正离子模式,多反应监测(MRM)模式;干燥气温度为325 ℃;干燥气流量为10 L/min;雾化气压力为137.9 kPa;毛细管电压为4 000 V。其他参数见表2。

表2 10种非甾体抗炎药的质谱参数Table 2 MS/MS parameters of ten non-steroidal anti-inflammatory drugs
*quantitative ion
2 结果与讨论
2.1 提取溶剂的优化
不同于液态样品的处理方式,肌肉等固态样品需先用溶剂提取后再进行分散液液微萃取。本实验考察了不同pH值(pH 3.5、5.0、7.5、9.5)的20 mmol/L醋酸铵缓冲溶液对马肉样品中10种药物的提取效率。结果显示,10种药物在酸性条件下的提取效率明显高于中性和碱性条件,这是由于非甾体药物中多含有N、O、S等杂原子和酰胺键、羧基键等,在酸性条件下易离子化,有利于提高化合物在水中的溶解度。文献[33]报道红细胞的存在会导致苯基丁氮酮氧化分解,使用10 mmol/L抗坏血酸可避免分解,本文最终选择提取溶剂为含10 mmol/L抗坏血酸的20 mmol/L醋酸铵缓冲溶液(乙酸调至pH 3.5)。
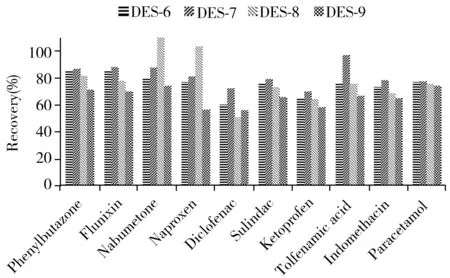
图1 不同萃取剂对NSAIDs萃取效率的影响Fig.1 Effect of various extraction agents on extraction efficiencies of NSAIDs
2.2 DES萃取剂的选择
氯化胆碱无毒安全、价格低,广泛用作DES的氢键受体[27]。本研究使用氯化胆碱按照表1合成了9种DES,按照“1.4”方法对10种目标物进行分散液液微萃取。结果显示,DES-1、DES-2、DES-3的水溶性较好,不能形成两相系统,无法萃取;DES-4、DES-5的黏度较大,不利于在水相中分散,操作难度大;以苯酚为氢键供体合成的DES-6、DES-7、DES-8、DES-9作为萃取剂时,可通过加入少量分散剂实现快速萃取,其中氢键受体和氢键供体比例为1∶2制得的DES-7萃取效率最佳(见图1)。进一步考察了DES-7用量(0.3、0.5、0.8、1.0 mL)对目标物萃取效果的影响,发现其用量为0.3 mL时难以收集DES层,测定结果不稳定;随着DES-7用量的增加,待测物的萃取效率均呈上升趋势,用量为0.8 mL时可获得较好结果,继续增加用量萃取效率无显著变化,因此选择DES-7萃取剂用量为0.8 mL。
2.3 分散剂的选择
常用分散剂包括乙腈、丙酮、四氢呋喃、甲醇等,考察结果显示乙腈、丙酮、四氢呋喃均能使DES萃取剂在水相中分散成细小液滴,增大萃取剂与待测物的接触面积。上述3种分散剂形成两相系统的分散能力:四氢呋喃>乙腈>丙酮,但对待测物的萃取效率:乙腈>四氢呋喃 >丙酮,综合考虑选择乙腈作为分散剂。考察了乙腈用量(0.3、0.5、0.8、1.0、1.5 mL)对目标物萃取效果的影响,发现萃取效率随乙腈体积的增加而增大,乙腈用量为0.8 mL时萃取效率达到最大值,此后则随乙腈用量的增加而减小。这是因为乙腈用量少时分散效果差,萃取剂未能均匀分散于水相中,导致萃取效率低;当乙腈用量过大时,会影响待测物在水相中的溶解度,导致萃取效率降低,因此实验选择分散剂乙腈的最佳用量为0.8 mL。
2.4 盐加入量的影响
氯化钠可通过盐析作用降低非甾体药物在提取液中的溶解度,同时还能促进萃取剂在水相中的分散,因此盐的加入能够促进待测物在DES萃取剂相的分配。考察了氯化钠加入量分别为0.25、0.5、1.0、1.5、2.0 g时10种非甾体抗炎药的萃取效率,结果表明,随着氯化钠加入量的增加,待测物的萃取回收率逐渐提高,其中舒林酸和吲哚美辛在氯化钠加入量为1.5 g时的回收率最高,其他8种待测物在氯化钠加入量为1.0 g时回收率最高。综合考虑选择氯化钠加入量为1.0 g。
2.5 基质效应
由于肌肉基质复杂,蛋白质、脂肪、磷脂和内源性代谢物等对待测物检测有显著干扰并影响结果的准确性[34]。采用提取后加入法计算绝对基质效应,公式为:基质效应(ME)=(空白基质中待测物质的响应值/乙腈中待测物质的响应值)×100%,ME>1为基质增强效应,ME<1为基质抑制效应。结果表明,苯基丁氮酮为中等强度的基质增强效应,ME=1.46;其余9种待测物为基质抑制效应,ME为0.40~0.71。为消除基质效应带来的定量偏差,本实验采用基质匹配标准工作溶液进行定量。
2.6 方法学考察
2.6.1 标准曲线及检出限在基质溶液中添加混合标准工作溶液,采用本方法进行测定,以各组分的峰面积为纵坐标(y),质量浓度为横坐标(x,ng/mL),在优化实验条件下考察了10种非甾体抗炎药的线性关系。结果显示,苯基丁氮酮、萘普生、托芬那酸、乙酰氨基酚的线性范围为5.0~100 ng/mL,其余6种药物的线性范围为0.1~10 ng/mL,相关系数(r2) 均大于0.996。根据3倍信噪比(S/N=3)计算检出限(LOD) ,10倍信噪比(S/N=10) 计算定量下限(LOQ),得到10种非甾体抗炎药的LOD为0.05~2.0 μg/kg,LOQ 为0.10~5.0 μg/kg(见表3)。
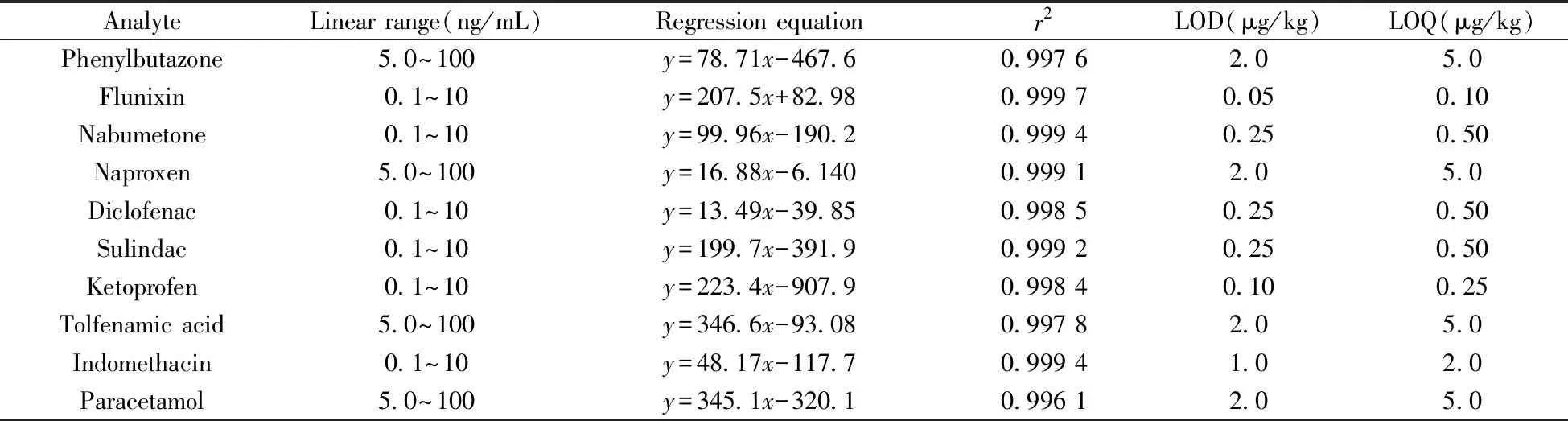
表3 10种NSAIDs的线性范围、线性方程、相关系数、方法检出限及定量下限Table 3 Linear ranges,linear equations,correlation coefficients(r2) ,the method detection and quantitation limits of ten analytes
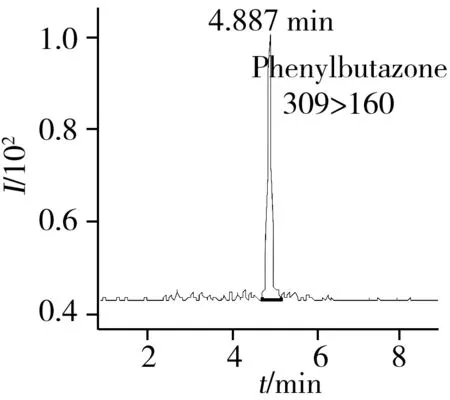
2.6.2 回收率与相对标准偏差选取阴性马肉样品,分别添加3个浓度水平的非甾体抗炎药混合标准溶液,混合均匀后按本方法进行前处理和检测,每个浓度水平平行测定6次,计算回收率和相对标准偏差(RSD)。表4结果显示,在5.0、10、25 μg/kg 3个加标水平下,10种NSAIDs的平均回收率为73.5%~94.6%,RSD为1.1%~ 8.1%,方法能够满足马肉中多种非甾体抗炎药的测定要求。图2为阴性马肉低水平(5.0 μg/kg)加标样品的MRM图。


2.7 实际样品检测
应用本方法对蒙古国进口5批冷冻马肉和农贸市场销售2份马肉样品进行检测,均未检出10种非甾体抗炎药。选取1个蒙古国进口冷冻马肉样品,按5.0 μg/kg水平添加NSAIDs混合标准溶液,按照本方法进行提取并测定,得到加标回收率为78.3%~93.2%,符合质控要求,方法可用于实际样品分析。

表4 马肉样品中10种非甾体抗炎药的平均回收率及相对标准偏差(n=6)Table 4 Average recoveries and RSDs of 10 NSAIDs in horse meat(n=6)
2.8 与标准方法的对比
将本方法与现行标准SN/T 2190-2008[9]和GB/T 20754-2006[10]方法进行比较,见表5。与传统的液液萃取法和固相萃取法相比,本方法的有机溶剂用量明显减少,大大降低了实验成本以及对环境和操作人员的危害;操作简单,萃取与净化一步完成,耗时短、效率高;检测灵敏度与标准方法相当或有所提高。

表5 本方法与标准方法的对比Table 5 Comparison between the method in this paper and the standard methods
3 结 论
本文建立了一种基于低共熔溶剂的分散液液微萃取/超高效液相色谱-串联质谱技术测定马肉中10种非甾体抗炎药的方法,以廉价易得的氯化胆碱和苯酚经过简单混合合成低共熔溶剂,减少了溶剂用量,节约了分析成本,降低了环境污染,且前处理操作简单快捷,在保证提取效率的同时缩短了萃取时间。方法检出限为0.05~2.0 μg/kg,平均回收率为73.5%~94.6%,RSD小于10%。该方法稳定可靠,满足动物源性食品质量监测的需求,为DES在兽药残留检测中的应用提供了新的思路。
猜你喜欢
中国保健营养(2019年6期)2019-10-21
科学导报(2019年19期)2019-09-23
学苑创造·A版(2019年8期)2019-08-15
食品与生活(2019年4期)2019-05-13
家庭用药(2017年7期)2017-07-29
医学美学美容·中旬刊(2015年2期)2015-10-21
环球时报(2013-02-18)2013-02-18
青年文摘·上半月(1983年9期)1983-01-01
