
m6A RNA甲基化调控因子对甲状腺癌进展及预后的影响
2021-05-21 02:28黄甜甜段碧晗叶迎春王硕高凌
疑难病杂志 2021年5期
黄甜甜,段碧晗,叶迎春,王硕,高凌
甲状腺癌是一种常见的内分泌系统恶性肿瘤,也是人类最常见的肿瘤之一。在世界许多地区,特别是发达国家,甲状腺癌的发病率呈上升趋势。分化型甲状腺癌在甲状腺癌中占90%以上,包括甲状腺乳头状癌和滤泡型甲状腺癌。然而,甲状腺癌通常是无症状的,芬兰一项经典研究发现,隐匿性甲状腺癌的患病率高达30%[1]。因此,了解甲状腺癌发病的分子机制对早期诊断、准确治疗及改善预后都至关重要。
N6-甲基腺苷(N6-methyladenosine,m6A)是一种甲基化修饰,可发生在RNA腺嘌呤上,是大多数真核生物最普遍的mRNA修饰形式[2]。在人类及其他许多物种中,m6A在调节基因表达、蛋白质翻译、细胞行为和生理状态等方面发挥着重要作用[3]。m6A甲基化是一个动态可逆的过程,根据13个m6A甲基化调控因子的功能不同,可以将其分为三类[4-5]:“书写者”介导RNA的甲基化修饰过程,包括METTL3、METTL14、KIAA1429、WTAP、RBM15和ZC3H13;“擦除者”介导RNA的去甲基化过程,包括FTO和ALKBH5;“阅读者”负责读取RNA甲基化信息,参与下游RNA的翻译和降解,包括YTHDC1、YTHDC2、YTHDF1、YTHDF2和HNRNPC。“擦除者”之一的FTO与人类肥胖和心理发展有关,而ALKBH5已被证明以去甲基化依赖的方式影响小鼠的精子发生[6-7],这表明在各种生理过程中m6A扮演着重要的角色。
越来越多的研究表明,m6A在许多复杂的人类疾病发生发展中发挥着重要作用[8-9]。关于m6A与胃癌、肝癌、乳腺癌、肺鳞状细胞癌等多种肿瘤疾病的关系已有较多报道[10-13],而m6A在甲状腺癌发生和进展过程中的作用仍不十分清楚。本研究利用TCGA数据库中的RNA测序数据,系统分析了13个广泛报道的m6A RNA甲基化调控因子在甲状腺癌中的表达。此外,使用LASSO Cox回归分析,筛选出3个m6A RNA甲基化调控因子构建LASSO风险回归模型,预测甲状腺癌患者的生存和预后,报道如下。
1 资料与方法
1.1 临床资料 从TCGA (https://cancergenome.nih.gov/)数据库中获取了甲状腺癌组织510例和正常甲状腺组织58例的RNA-seq转录组数据及相应甲状腺癌患者临床病理数据。在分析甲状腺癌亚组的总体生存率和临床病理因素时,排除了缺少年龄、性别、病理分级和随访数据的样本,最终保留了501例甲状腺癌样本,其中甲状腺癌患者中男314例,女187例;年龄≤60岁171例,>60岁330例;生存状态:死亡182例,存活319例;临床分级:1级36例,2级184例,3级281例;临床分期:Ⅰ期79例,Ⅱ期152例,Ⅲ期205例,Ⅳ期65例;原发肿瘤:T139例,T2111例,T3215例,T4136例;远处转移:M0451例,M150例;淋巴结转移:N0153例,N1138例,N2104例,N3106例。
1.2 生物信息学分析方法 主要使用Active-Perl 5.24.3 Build 2404(https://www.perl.org)、Rv3.6.0 (https://www.r-project.org/)软件对数据进行转换、统计分析和计算。使用R软件包中的“pheatmap” 软件包比较甲状腺癌组织和正常甲状腺组织中13个m6A RNA甲基化调控因子的表达。使用“Consensus Cluster Plus” 软件包通过一致性聚类分析方法将甲状腺癌组织分为2个亚组,探究m6A RNA甲基化调控因子在甲状腺癌中的作用。主成分分析“PCA”包检测基于m6A相关基因表达的肿瘤分组的可行性。为了探讨m6A RNA甲基化调控因子的预后价值,使用Lasso Cox回归分析方法来确定与总生存率显著相关的3个基因,并得到最终风险评分公式。根据每位患者的风险评分,将其分为高危组和低危组,并使用R包中的“survival”“survminer”“pheatmap”软件包比较2组总体生存率和临床病理特征。使用2组间的ROC曲线来检验生存模型的预测效率,并使用单因素和多因素Cox回归分析方法确定风险评分的预后价值。
1.3 统计学方法 主要采用R软件对数据进行统计分析。以Wilcoxon秩和检验比较m6A RNA甲基化调控因子在肿瘤组织和正常组织中的表达情况。根据m6A RNA甲基化调控因子的表达情况将患者分为2个亚组后,采用卡方检验比较聚类 1和聚类2的性别、年龄、WHO分期和病理分级。在总体生存分析中,采用Kaplan-Meier法检测甲状腺癌亚组间、高危组和低危组间的总体生存率。使用ROC曲线分析评估风险评分对5年生存率的预测效率。通过单因素和多因素Cox回归分析确定风险评分的预后价值。P<0.05为差异有统计学意义。
2 结 果
2.1 m6A RNA甲基化调控因子在甲状腺癌中的表达情况 分析TCGA数据库中501例甲状腺癌样本和58例正常甲状腺组织样本中13个m6A RNA甲基化调控因子的表达情况。除YTHDF2外,12个调控因子均表现出显著的差异表达(图1A、B)。与正常组织比较,甲状腺癌组织的METTL3、YTHDC2、WTAP、ALKBH5、YTHDF1、METTL14、YTHDC1、FTO、KIAA1429、ZC3H13、RBM15显著下调(P<0.001),HNRNPC显著上调(P<0.001)。为进一步了解13个m6A RNA甲基化调控因子之间的关系,构建了PPI网络图来结果显示,WTAP、HNRNPC和YTHDC1似乎处于中心位置,并与其他m6A RNA甲基化调控因子相互作用 (图1C)。此外,大多数m6A RNA甲基化调控因子都显示出正相关关系。其中,ZC3H13与KIAA1429、KIAA1429与METTL14、METTL14与YTHDC1等3对调控因子呈较强的正相关关系(ρ=0.78),只有HNRNPC和ALKBH5呈负相关关系(ρ=-0.18)(图1D)。
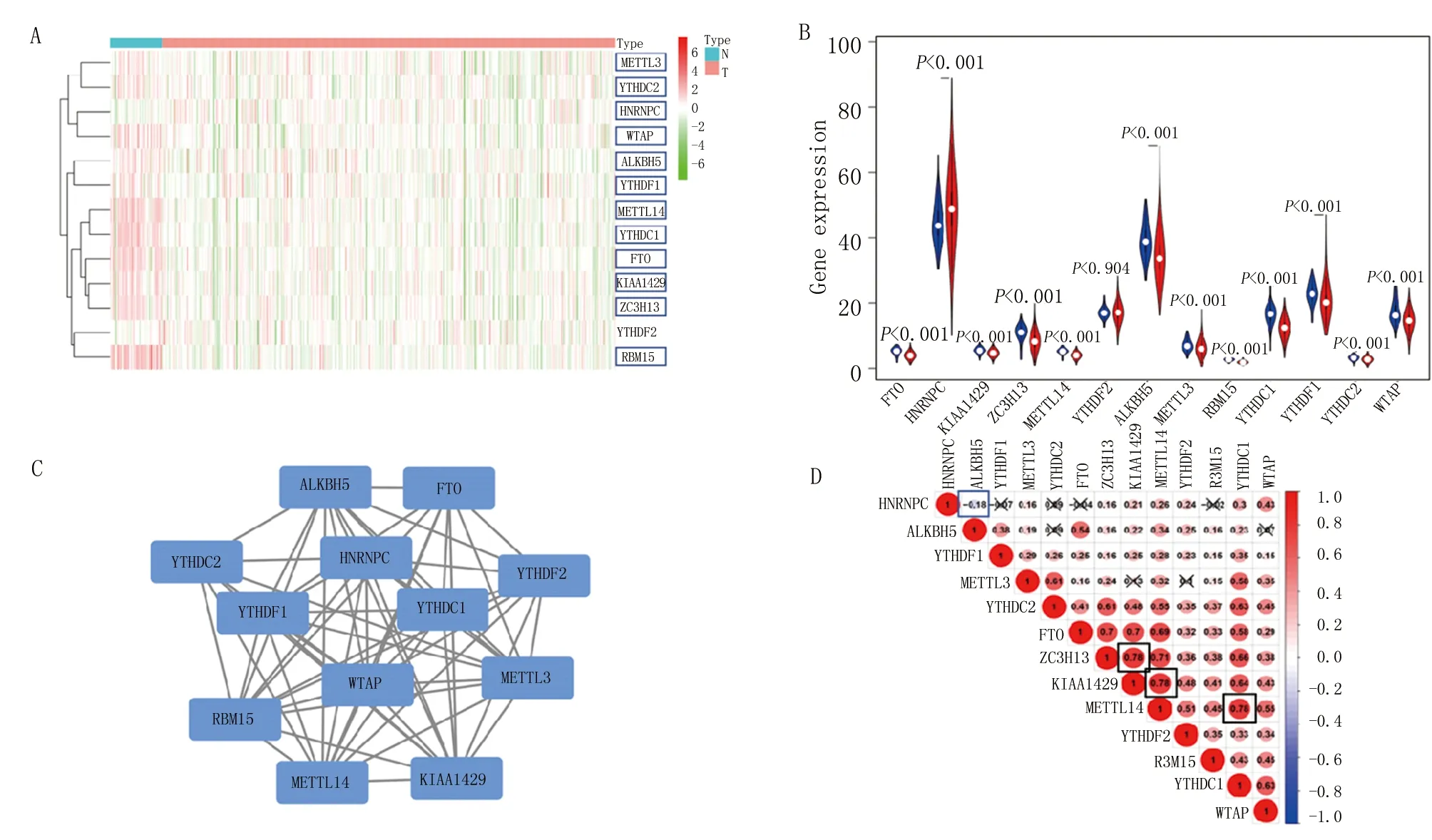
注:A.甲状腺癌与正常甲状腺组织中13个m6A RNA甲基化调控因子的基因表达水平(蓝色代表正常组织,粉红色代表甲状腺癌组织)。红色越深表示基因表达上调越明显,绿色越深代表基因表达下调越明显;B.小提琴图可视化m6A RNA甲基化调控因子在甲状腺癌中的表达差异;C.PPI网络图评估m6A RNA甲基化调控因子之间的相互作用;D.Spearman相关分析每个m6A RNA甲基化调控因子之间的关系
2.2 m6A RNA甲基化调控因子的一致性聚类分析 采用一致性聚类分析对501例甲状腺癌组织进行分组,分组标准如下:(1)累积分布函数增长率较慢;(2)聚类后各组样本量足够大;(3)组内相关性高而组间相关性低。在TCGA数据集中,根据m6A RNA甲基化调控因子的表达相似性,最终将甲状腺癌组织分为2个亚组(图2A~C)。对甲状腺癌2个亚组的主成分分析验证表明,总RNA在亚组中的表达具有特异性,即聚类1似乎有向中心聚集的趋势,而聚类2则有向四周扩散的趋势(图2D),这说明根据m6A RNA甲基化调控因子的一致性聚类分析可以将甲状腺癌很好地区分为2个亚组。但2个亚组之间存在很多重叠的区域,这表明甲状腺癌2个亚组之间具有一些共同点。

注:A.k = 2~9的一致性聚类累积分布函数;B.k =2~9时累积分布函数曲线下面积的相对变化;C.当k = 2时,一致性聚类分析将TCGA甲状腺癌数据分成2个不同的亚组;D.TCGA数据集总RNA表达谱的主成分分析,红色代表聚类1,蓝色代表聚类2
2.3 聚类结果与临床转归及临床病理特征的关系 进一步分析甲状腺癌患者的总体生存率及临床病理特征发现,聚类1的总体生存率明显高于聚类2(图3A)。聚类1的4年生存率约为97%,聚类2约为87%。此外,在聚类1中HNRNPC明显高表达,而ALKBH5的表达明显降低,进一步说明这两种调控因子之间的负相关关系。与聚类 1比较,聚类 2与男性、高临床分期和高T状态显著相关(图3B)。总而言之,聚类结果与甲状腺癌患者的总体生存率及临床肿瘤恶性程度密切相关。
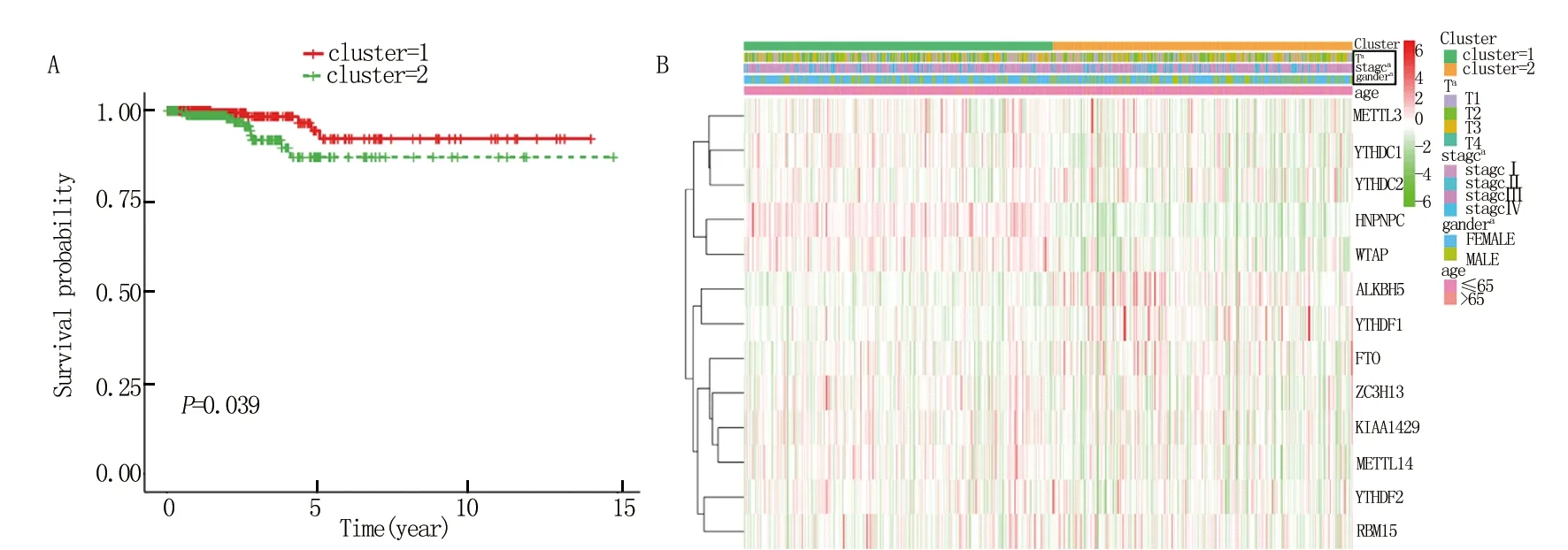
注:A.聚类1总体生存率明显高于聚类2(P<0.05);B.聚类 1和聚类 2临床病理特征比较差异有统计学意义(P<0.05);与聚类1相比,aP<0.05,bP<0.01
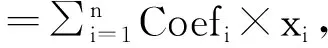
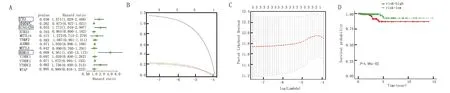
注:A.TCGA数据集中与总体生存率相关的13个m6A RNA甲基化调控因子的单变量Cox回归分析,根据风险比、95%置信区间确定了3个基因(P<0.05,HR>1);B、C.在TCGA数据集中选择靶基因的过程;D.根据风险评分将甲状腺癌患者分为高危组和低危组的总体生存曲线
2.5 甲状腺癌危险评分与预后及临床病理特征的关系 ROC曲线结果表明,风险评分对甲状腺癌患者的5年生存率有较高的预测准确率(AUC=0.717)(图5A)。为进一步了解高危组与低危组甲状腺癌患者3个m6A RNA甲基化调控因子的表达情况及其临床病理特征的关系,再对不同分组与临床病理特征(性别、年龄、生存状态、T状态)进行相关性分析并绘制其m6A RNA甲基化调控因子的表达热图(图5B)。结果显示,与低危组比较,高危组RBM15、KIAA1429、FTO表达明显增高,且高危组和低危组甲状腺癌患者在T状态之间存在显著差异。
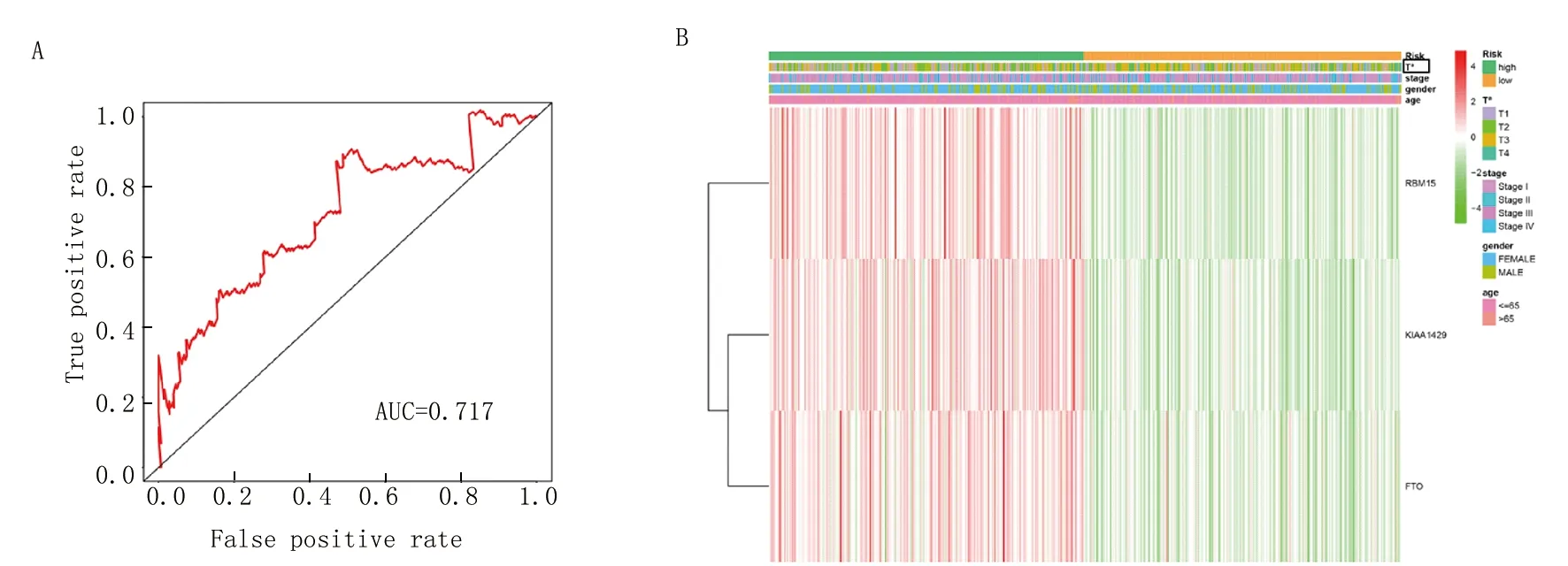
注:A.ROC曲线显示风险评分的5年生存率预测价值;B.热图显示3种m6A RNA甲基化调控因子在甲状腺癌患者高危组和低危组的表达水平以及临床病理特征差异;与甲状腺癌患者高危组相比,aP<0.05,bP<0.01
2.6 甲状腺癌的独立预后指标分析 单因素Cox回归分析显示,年龄、风险评分、分期、T状态均与总体生存率相关。多因素Cox回归分析结果显示,年龄和风险评分可以作为甲状腺癌的独立预后指标,即甲状腺癌的风险随着年龄和风险评分的增加而增加,见图6。
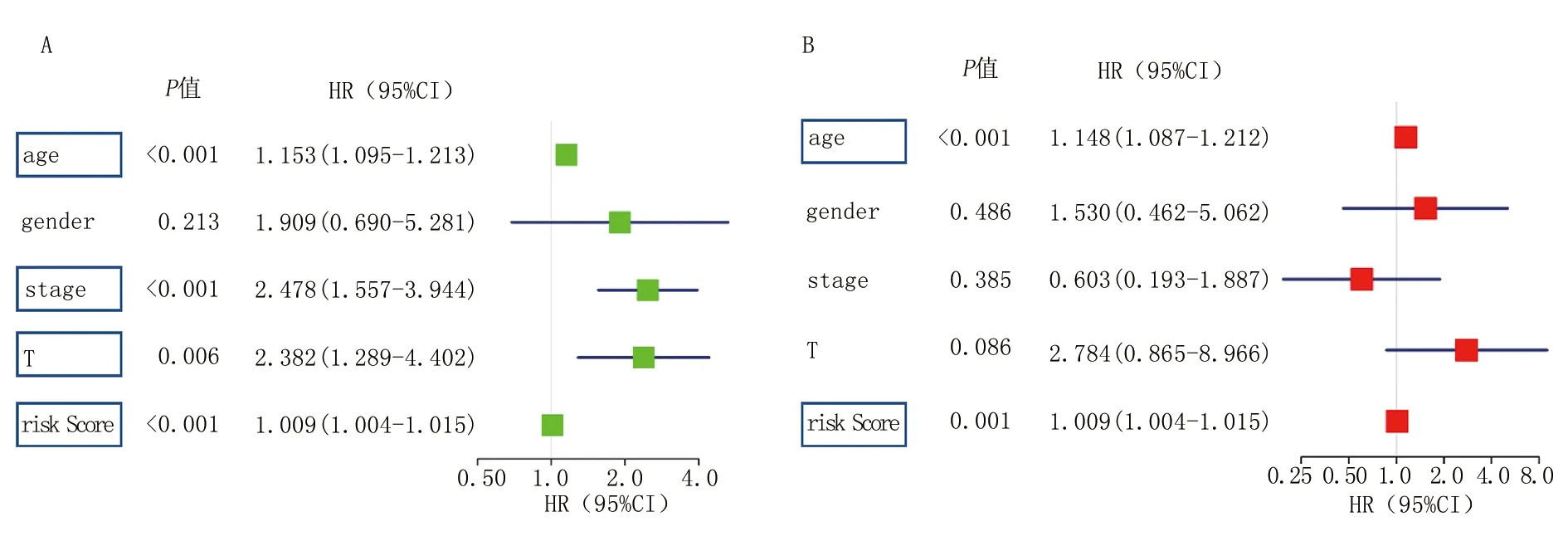
注:A.TCGA数据集中临床病理因素和风险评分与患者总体生存之间的单变量Cox回归分析;B.TCGA数据集中临床病理因素和风险评分与患者总体生存之间的多变量Cox回归分析
3 讨 论
甲状腺癌是最常见的内分泌系统恶性肿瘤,近40年来在世界范围内发病率呈上升趋势。超声检查的进步可能是造成这种趋势的原因[14],然而,导致这一增长的其他原因尚不清楚。虽然甲状腺癌相对于其他恶性肿瘤具有较好的惰性和良好的长期生存率,但甲状腺癌可发生在任何年龄,通常对年轻人的影响更大[15]。m6A RNA甲基化调控因子在肿瘤的发生发展中起重要作用,且已被证实与多种癌症密切相关,靶向m6A RNA甲基化的抗癌药物在临床的应用前景非常广阔[16]。因此,有必要探讨m6A RNA甲基化调控因子与甲状腺癌的关系或对甲状腺癌预后的预测价值。
在本研究中,首先分析了m6A RNA甲基化调控因子在甲状腺癌和正常甲状腺组织中的表达情况及其与不同临床病理的关系。甲状腺癌组织和正常组织中多达12个m6A RNA甲基化调控因子的表达比较差异有统计学意义。
急性髓系白血病患者中METTL3表达升高,并发挥致癌作用。METTL3通过增加cMYC、BCL-2和PTEN的翻译,抑制细胞分化和凋亡,促进细胞增殖。MELLT3还激活PI3K / AKT通路,控制细胞分化和自我更新[17]。肝细胞癌中METTL3表达升高,METTL3在体外可促进肝癌细胞的生长、迁移和集落形成,在体内可影响肝癌的成瘤性、生长和肺转移,且METTL3水平升高提示预后不良[16]。METTL3在卵巢癌中也高表达,且METTL3高表达与肿瘤分级、FIGO分期及总生存率显著相关[18]。近期,有学者进行了METTL3促进甲状腺癌进展的相关研究[19],研究表明,METTL3诱导TCF1 mRNA m6A甲基化,上调TCF1蛋白水平,TCF1通过激活Wnt通路进一步刺激肿瘤细胞转移,从而加重甲状腺癌的恶性进展,提示METTL3是一种潜在的癌基因。然而,本研究显示,METTL3在甲状腺癌患者中的表达远低于正常组织,提示METTL3可能阻碍了甲状腺癌的进展。这表明METTL3在甲状腺癌发生发展中的作用可能还需要更多的研究来证明。
在探索m6A RNA甲基化调控因子是否对甲状腺癌有预后价值的过程中,进行了Cox单变量分析和LASSO回归分析,鉴定筛选出RBM15、KIAA1429和FTO等3个基因。研究发现,RBM15、KIAA1429和甲状腺疾病的关系研究甚少,而FTO与甲状腺癌密切相关。
RBM15对基因敲除模型小鼠中多种组织的发育至关重要,尤其对维持造血干细胞长期稳态和巨核细胞、B细胞的分化具有重要作用[20]。此外,RBM15参与了染色体易位t (1;22),产生与急性巨核细胞白血病相关的RBM15-MKL1融合蛋白[21]。除参与急性巨核细胞白血病的发生外,RBM15还是X染色体沉默的重要因素,并已证实在乳腺组织中表达[22]。研究表明,雌激素可独立增加RBM15的表达,并与乳腺疾病的发生有关。对于甲状腺癌,女性的发病率高于男性,这是否与雌激素信号传导有关还有待进一步研究。
KIAA1429已被证实通过不同途径影响多种癌症,包括通过依赖于GATA3的m6A转录后修饰促进肝细胞癌的进展,以及通过直接靶向c-jun mRNA调节胃癌细胞增殖[23]。此外,一项关于乳腺癌的研究表明,KIAA1429可通过调节CDK1表达而成为乳腺癌的致癌因子[24]。各种CKD抑制剂被广泛应用于临床治疗恶性肿瘤,包括甲状腺癌[25]。因此,KIAA1429与甲状腺癌的关联机制值得进一步探讨。
FTO在乳腺癌、急性髓系白血病和胶质母细胞瘤中高表达,并促进这些癌症的进展,现阶段已开发出相关药物[10,26-27]。大黄酸是第一种有效的FTO抑制剂,通过竞争性结合催化区域和单链RNA底物抑制FTO。大黄酸还能有效抑制m6A去甲基化,提高体外m6A水平[28]。然而,本研究发现FTO在甲状腺癌中表达水平较低。此外,它在膀胱癌中也呈低表达。m6A调控基因的致癌作用似乎存在争议。这可能是由于癌症的异质性,相同的基因在不同的癌症中扮演不同的角色,导致m6A调节基因在不同癌症中的表达差异。FTO更多的是作为一个与肥胖、糖尿病和代谢综合征相关的基因进行研究,在一些大型的研究中发现,BMI增加已成为甲状腺癌一个新的危险因素。
综上,m6A RNA甲基化调控因子在甲状腺癌中异常表达并具有预后价值,为进一步探讨m6A甲基化在甲状腺癌中的作用和寻找相关治疗靶点提供了重要证据。
利益冲突:所有作者声明无利益冲突
作者贡献声明
黄甜甜:设计研究方案,实施研究过程,论文撰写;高凌:提出研究思路,分析试验数据,论文审核;段碧晗:实施研究过程,资料搜集整理,论文修改;叶迎春:进行统计学分析;王硕:课题设计,论文撰写
猜你喜欢
中国合理用药探索(2022年7期)2022-08-16
上海师范大学学报·自然科学版(2022年3期)2022-07-11
岭南心血管病杂志(2022年3期)2022-06-27
中国农学通报(2022年13期)2022-05-31
检验医学与临床(2022年5期)2022-03-21
昆明医科大学学报(2022年1期)2022-02-28
农学学报(2021年11期)2021-12-12
中国烟草科学(2021年6期)2021-01-09
右江医学(2019年11期)2019-12-27
中西医结合心血管病电子杂志(2018年17期)2018-09-12
