
炎症在颅内动脉瘤破裂中作用的研究进展
2021-05-20 09:24:30王介南朱悦琦
介入放射学杂志 2021年4期
王介南, 朱悦琦
非创伤性蛛网膜下腔出血患者中近85%是由颅内动脉瘤(intracranial aneurysm, IA)破裂引起,其导致的死亡率超过了25%[1-2]。 IA 在破裂前可无任何症状, 研究显示无症状的IA 患者是已破裂IA患者数量的10 倍,更需要引起临床重视[3]。 IA 的病理特征在于由血管内皮功能障碍、内膜增生、细胞外基质(ECM)的破坏和炎性反应引起动脉壁完整性的丧失[4]。 炎症在这其中具有较强的促进作用[5],多个研究表明,炎性反应在对动脉壁完整性的破坏中起重要作用[6-8]。炎性细胞浸润在动脉瘤不同阶段的是一个持续发展的过程[9]。炎性反应相关过程,如基质金属蛋白酶(matrix metalloproteinase, MMP)破坏弹性纤维、 人单核细胞趋化蛋白-1(monocyte chemoat-tractant protein-1,MCP-1)和转录因子Ets-1(E26 transformation-specific-1) 蛋白诱发巨噬细胞浸润,和巨噬细胞致平滑肌细胞(SMC)缺失并释放MMP 等,均在引起IA 破裂中扮演了重要角色。可以发现,引发炎症的细胞和因子之间有间接或直接的联系,并通过相关的蛋白通路和基因调控影响着IA的破裂。
IA 破裂是一个多因素导致的结果,而炎症可直接引起IA 壁退化并导致IA 增大, 是最终增加IA破裂风险的关键环节[9]。 IA 瘤壁在炎性作用下持续再塑形失代偿而不能抵抗血流动力学的应力时,便会引起破裂[4,10]。 现阶段能够预防IA 进展和破裂的药物疗法仍在研究中,这些研究大多是基于炎症在IA 中的作用通路。 炎症与IA 破裂机制及其抗炎治疗的相关研究将为IA 的临床预防和治疗提供新的选择, 同时对于改善IA 破裂后的临床预后具有积极作用。 本文就炎症与IA 破裂关系,及其可能的抗炎治疗降低IA 破裂风险进行综述。
1 炎性细胞因子与IA 的破裂
在动脉瘤早期,如高动脉剪切应力引起的血管内皮损伤导致血管内皮功能障碍,包括血管内分泌功能紊乱、抗血栓形成作用降低、调节血管张力受限等。血管内皮功能障碍促使内皮细胞的数量减少并使部分SMC 暴露于血液中从而激活了转录因子Ets-1 的表达。 Ets-1 可介导IA 瘤壁中MCP-1 的表达并招募更多的巨噬细胞从而促进IA 的进展[7]。巨噬细胞又可直接促进其他炎性细胞因子的释放,包括MMP、TNF-α、IL-1β 等[11]。已有研究表明,人主动脉瘤中的IL-1β 蛋白水平比正常主动脉高约20倍,其可增加MCP-1 的产生并加剧炎性反应以促进主动脉瘤的进展[12],但IL-1β 在IA 中的具体作用有待进一步研究。 炎性细胞因子活跃在IA 的各个阶段,在破裂时作用尤为突出。
1.1 MMP 与IA 破裂
MMP 是一种可以降解细胞外基质的蛋白水解酶[13],在动脉瘤中可以检测到MMP 的表达增加[14]。MMP-2 和MMP-9 的基因表达在破裂动脉瘤中明显高于未破裂的动脉瘤[15]。
金属蛋白酶组织抑制剂(TIMP)被认为是MMP在组织中的关键抑制因子。 在人类IA 中可以检测到丰富的TIMP 表达。 TIMP-1,TIMP-2 和TIMP-3的表达在破裂动脉瘤中高于未破裂动脉瘤[16]。在近期 研 究 中,Kimura 等[17]在 研 究25 例 主 动 脉 夹 层 患者时,发现TIMP-3, TIMP-4 的表达降低。 这可能与miR-21-5p 的表达增加抑制了TIMP-3 的表达,最终导致MMP-TIMP 平衡失调,并引起主动脉夹层的破裂。综上,动脉瘤破裂与MMP-2、MMP-9、TIMP 的表达密切相关, 而MMPs 和TIMP 之间的失衡可能在破裂中起重要作用[18-19]。
脂多糖(LPS)是革兰阴性细菌细胞壁的一部分,已有研究表明LPS 可促进IA 破裂[20]。 其作用途径可能通过刺激Toll 样受体4(Toll-likereceptor 4,TLR4)激活核因子κB(NF-κB)家族的表达来实现[21]。LPS/TLR4/NF-κB 信号通路参与了免疫相关细胞因子TNF-α 的表达[22]。TNF-α 和IL-1β 等促炎性细胞因子诱导MMP 的前体(proMMP)变为MMP。 最终,MMP 通过LPS/TLR4/NF-κB 信号通路被激活以降解ECM 和紧密连接蛋白[23],导致IA 壁脆性增加(如图1)。
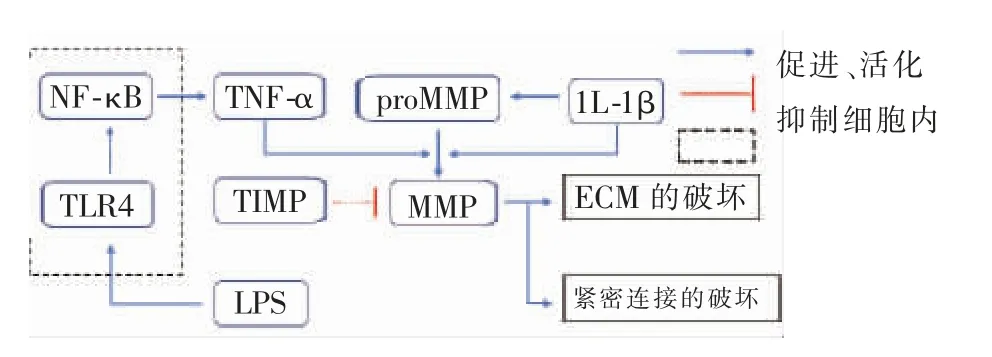
图1 LPS/TLR4/NF-κB 信号通路激活MMP
研究发现炎性因子如MMP-9 可以降解弹性蛋白,而弹性蛋白是IEL 的蛋白质成分[19]。 Chatzizisis等[24]发现内皮细胞的减少,炎性细胞的积聚和MMP的活性增加可导致IEL 的严重破坏。 在15 个人类破裂IA 样本的检测中,也没有观察到IEL[25]。 MMP活性增加导致IEL 的减少,而做为血管壁弹性结构的IEL 的丧失增加了IA 的破裂风险。
1.2 TNF-α 与IA 破裂
TNF-α 可通过引起内皮细胞功能障碍,血小板聚集以及血管SMC 功能和增殖的改变以介导血管功能障碍。TNF-α 在动脉瘤的形成和破裂中均起到了关键作用[26],且TNF-α 与IA 破裂风险增加相关[6]。 巨噬细胞产生TNF-α 可提高细胞凋亡相关蛋白的活化[27],使IA 的管壁变薄。 而TNF-α 是IA 中关键的炎性反应因子, 可通过介导内皮损伤、SMC 表型转变、细胞趋化因子的激活、基质重塑基因的上调、自由基的产生导致氧化应激,并最终引起细胞凋亡[28]。
2 炎性细胞与IA 的破裂
多宗研究已经证实,IA 中的炎性细胞主要是巨噬细胞和T 淋巴细胞[7],和少量B 淋巴细胞,其浸润的程度被认为与IA 破裂有关[29]。 T 细胞是人体细胞免疫的关键。 Frosen 等[29]学者证明在破裂的IA 壁中浸润的T 细胞数量要大于未破裂的IA。 在Miyata 等[30]的研究中,虽然在IA 壁中检测到T 细胞,但相对于巨噬细胞介导的炎症和管壁退行性改变导致IA而言,T 细胞的作用较弱。 故T 细胞在IA 发病机理中的作用还有待阐明。
巨噬细胞是渗透IA 病变的主要炎性细胞,它的积聚可进一步放大和介导炎症[31]。 NF-κB 在巨噬细胞中被激活并诱导MCP-1 和血管细胞黏附分子1(vascular cell adhesion molecule-1, VCAM-1)的表达[32]。MCP-1 和VCAM-1 都是参与单核细胞调控的刺激成分,可进一步募集巨噬细胞,导致其浸润到IA 壁的数量不断增加, 在IA 破裂时浸润的程度更为突出[29,33]。 Hosaka 等[34]发现小鼠IA 模型破裂比例增大时,巨噬细胞浸润明显增多。 他们认为巨噬细胞和动脉瘤破裂还存在一定数量依赖的关系。Korkmaz 等[35]通过显微手术方式切除6 个未破裂和6 个破裂的人类IA 标本,研究后发现巨噬细胞在破裂的IA 中增加明显, 其浸润到动脉瘤壁导致SMC的缺失并释放MMP 导致细胞外基质的降解, 最终诱导IA 的破裂。
研究发现,IA 破裂与血管壁细胞数量减少有关[7]。SMC 作为颅内动脉重要的组成细胞,在生理条件下,可通过舒张和收缩调节血管的张力。 IA 壁退化过程中,很重要一部分是细胞缺失,尤其是SMC的缺失。 内皮细胞和SMC 的缺失被Frosen 等[29]认为是动脉瘤破裂的标志。这可能是因为SMC 合成的细胞外基质保持了IA 壁的结构稳定性。 在正常的大脑动脉壁中,SMC 主要是收缩表型[36],能够精细调节血流量。 然而,在IA 中,部分SMC 经历了从收缩表型向合成表型的调节转变[37]。 这些转变导致SMC 合成的细胞外基质减少且细胞代谢时间延长,从而导致IA 壁易破裂性增加。
此外,Frosen 等[29]发现IA 中SMC 排列规则时,动脉壁稳定性好,破裂风险降低;而当SMC 排列散乱时,动脉壁稳定降低,破裂风险增高。SMC 的排列散乱可能与巨噬细胞浸润引起SMC 数量减少有关,而SMC 数量减少又可导致IA 壁的纤维化。 纤维化通常继发于炎性细胞侵袭,被认为是慢性炎性反应的终点阶段[38]。 纤维化主要病理改变为器官组织内纤维结缔组织增多,实质细胞减少。 破裂的IA 壁常表现为严重或完全的纤维化。 故巨噬细胞浸润引起SMC 数量减少, 进一步引起IA 壁稳定性降低及纤维化最终导致IA 的破裂。 综上,炎性因子募集炎性细胞,炎性细胞又放大了炎性反应,这种级联式的反应促进了IA 的增长并引起破裂。
3 其他炎症因素与IA 的破裂
3.1 基因调控
Kurki 等[39]的 研 究 中,破 裂 的 囊 状IA 壁 中 有686 个基因上调,740 个基因下调。 上调的基因调控的生物过程包括对血流动力学的响应、白细胞的迁移、 氧化应激反应、 血管重塑和细胞外基质降解。miRNA 是一类由内源基因编码的长度约为22 个核苷酸的非编码单链RNA 分子, 它们参与转录后的基因表达调控。 Chen 等[40]研究认为miRNA 在囊状IA 破裂中可能起关键作用。 与此同时,Jiang 等[41]研究了14 例破裂IA 病例,发现18 种miRNA 的表达显着下调,且这些miRNA 都在SMC 中高度表达。随后,Sun 等[42]研 究 显 示IA 患 者 的 血 清miR-29 b 水平显着低于正常受试者, 且破裂组的miR-29 b 水平低于未破裂组。 miR-29b 能抑制NF-κB 信号传导的负调节因子TNFAIP3 的表达[43]。IA 中的miR-29 b减少,可能使NF-κB 信号通路表达增加。 NF-κB 表达可介导巨噬细胞活化使SMC 数量减少,从而促进IA 破裂。
此 外,Xin 等[44]利 用 小 鼠 模 型 还 发 现miR-143和miR-145 可通过一系列调节来阻断SMC 表型转换。 正常小鼠主动脉富含miR-143 和miR-145。 在缺乏miR-143 和miR-145 的小鼠中,SMC 对血管内皮损伤没有形成新内膜。 综上,miRNA 参与IA 的破裂, 且miRNA 的变化对IA 的破裂也有预警作用[42]。近年更多的关于动脉瘤的基因研究也为IA 的基因靶向治疗提供了理论依据[45]。
3.2 骨桥蛋白(osteopontin, OPN)
OPN 广泛分布于多种组织和细胞中,能够参与瘢痕组织的修复及自身代谢等。OPN 可由多种炎性细胞产生,并被证实可以上调MMP-2 和MMP-9 的活性[46]。OPN 的表达被认为可在血管炎症中起关键作用[47]。 在兔囊状动脉瘤模型中,OPN 的表达在高长宽比的动脉瘤中更常见[46],而高长宽比的动脉瘤形状在临床中更容易破裂。
3.3 髓过氧化物酶(myeloperoxidase, MPO)
在Gounis 等[48]的研究中,中性粒细胞中富含的MPO 和IA 的破裂也存在一定关系,这可能和MPO介导的持续炎症相关。 Ollikainen 等[49]学者利用36个人类动脉瘤标本分析MPO 与IA 的关系, 发现MPO 在破裂的IA 中广泛表达。 MPO 来源于嗜中性粒细胞,并导致SMC 的缺失,认为MPO 是IA 破裂的独立危险因素。
4 药物在IA 破裂中的作用
4.1 抗炎药
4.1.1 阿司匹林Hasan 等[50-51]学者的研究表明阿司匹林可能在IA 的进展和破裂中起保护作用。 他们利用巨噬细胞作为炎症替代标记物, 发现了每日服用阿司匹林的IA 患者3 个月后可以减轻IA 炎症的影像学证据。当然,上述基础疾病较多病例,其破裂风险降低也可能部分归咎于其他高危因素(如血压)的严格控制。 已知阿司匹林抑制MMP-2 和MMP-9[52]、COX-2 的表达[53]以及SMC 中的TNF-α的释放, 并降低NF-κB 活性来抑制炎性细胞黏附。Li 等[54]也 通 过Sprague-Dawley 大 鼠 实 验 得 出 了 阿司匹林通过抑制巨噬细胞的浸润而减缓动脉瘤的进展, 从而得出阿司匹林可能在IA 破裂中起保护作用。
4.1.2 多西环素 多西环素是一种抗微生物药物,它在体外和体内对MMP 活性抑制效果显著[55]。 在Nuki 等[56]的实验中,经过多西环素处理小鼠的IA发病率降低,在Makino 等[57]学者的实验中,在动脉瘤发率病一定的情况下, 经过多西环素处理小鼠的IA 破裂概率下降。 虽然多西环素直接应用于人体预防或治疗IA 还存在诸多问题, 但通过抑制MMP 活化来降低IA 的破裂的风险是颇具前景的治疗途径。
4.2 炎性因子抑制剂
4.2.1 前列腺素E 受体2(EP2)拮抗剂 EP2 信号传导参与内皮细胞和巨噬细胞中的NF-κB 活化[31]。在IA 壁中的SMC 中可以检测到EP2 的表达[58]。Aoki 等[31]发现EP2 可在IA 壁的全层被检测到;使用EP2 拮抗剂对患有IA 的大鼠进行处理后发现巨噬细胞浸润以及IA 的形成和进展都受到了抑制。
4.2.2 TNF-α 抑制剂 有证据支持TNF-α 在脑IA发病机制中的促进作用, 而不仅仅是IA 形成和破裂的炎症后副产物。Yokoi 等[59]的实验中,给予TNF-α抑制剂的大鼠血管中膜厚度显着增加并且动脉瘤大小减小。 TNF-α 抑制剂可抑制MMP-9 和诱导型一氧化氮合酶(inducible nitric oxide synthase, iNOS)的表达。 TNF-α 抑制剂的使用显著延缓大鼠动脉瘤的进展。 Starke 等学者通过实验也得出了相似的结论,TNF-α 抑制剂可有效预防动脉瘤进展和破裂[26]。TNF-α 抑制剂的安全有效性及剂量和起效时间仍需进一步研究[28]。
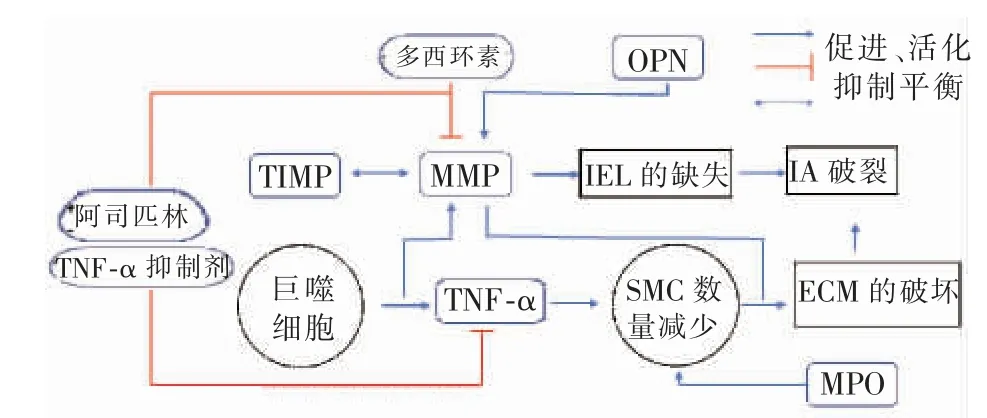
图2 炎症在IA 破裂中作用的模式图
综上所述,与炎症相关的因子和炎性细胞彼此的级联式放大作用, 导致了动脉瘤管壁的逐步退化,并最终引起破裂(如图2)。 MMP 促使细胞外基质降解,巨噬细胞和TNF-α 介导的动脉壁炎性反应等都在IA 破裂中发挥了重要作用。 作为无创药物疗法难以干预的疾病,IA 的破裂伴随的不可预测的灾难性后果,仍然是全球医学面临的挑战。 因此,更好地了解IA 破裂中炎症过程的相关机制和信号通路可能有助于建立有效的药物疗法, 以预防IA 破裂和导致的蛛网膜下腔出血。 每一个炎性因子都可能在破裂过程中产生影响,它们对动脉瘤的作用的比重也是一个有意义的话题。 这些炎性因子的研究将为IA 患者预防和治疗提供一种新的方式。
猜你喜欢
感染、炎症、修复(2021年1期)2021-07-28 06:18:10
天津医科大学学报(2021年3期)2021-07-21 09:03:48
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:30
上海农业学报(2017年3期)2017-04-10 12:39:26
医学研究杂志(2015年9期)2015-07-01 17:28:19
中国当代医药(2015年16期)2015-03-01 02:03:13
遵义医科大学学报(2014年1期)2014-08-09 01:29:38
中国药理学通报(2014年2期)2014-05-09 08:22:39
中医研究(2013年3期)2013-03-11 20:26:39
金属矿山(2013年11期)2013-03-11 16:55:05
