
水稻苗期抗稻瘟病基因全基因组关联分析
2021-05-15 09:27陈汝斌彭沙莎康厚祥
激光生物学报 2021年2期
陈汝斌,彭沙莎,王 丹*,康厚祥
(1. 湖南农业大学农学院,长沙 410128;2. 中国农业科学院植物保护研究所/植物病虫害生物学国家重点实验室,北京 100193)
水稻(Oryzae sativaL.)是世界上最重要的粮食作物之一,养育了世界近一半以上的人口[1]。据研究,到2050 年,平均每年需要增加44 万t 以上的粮食产量以满足全球的粮食需求[2]。由稻瘟菌(Magnaporthe oryzae)引起的稻瘟病是一种长期侵害水稻的真菌病害,几乎发生在水稻生长发育的整个时期[3]。根据侵染部位可将其分为苗瘟、叶瘟、穗颈瘟、节瘟以及谷粒瘟等类型[4]。每年因稻瘟病造成的水稻收成损失可达到10%~30%,严重时达50%以上,甚至颗粒无收,给我国的粮食生产及稻米品质造成了严重的损失。目前针对稻瘟病发生的防止措施是化学药剂防治以及培育优良广谱抗性品种。相比于前者,后者具有经济、环保、高效等特点[5]。然而,由于田间稻瘟菌生理小种基数大、传播和变异速度快等原因,已有抗病基因的水稻其抗性在推广3~5 年后容易丧失,因此快速挖掘新的稻瘟病抗性基因对培育抗稻瘟病品种具有重大的意义[6]。
目前,定位复杂性状主要有两种方式,一是经典的连锁分析,二是关联分析。连锁分析基于双亲杂交构建遗传作图群体,通过遗传标记对个体进行基因分型,并对结果进行分析。但该方法存在构建群体耗费时间长且每个位点只能鉴定2 个等位基因等缺点,因此所鉴定数量性状位点(quantitative trait locus,QTL)的数量受到限制[7]。而关联分析相较于连锁分析有以下几个优点:1)目标群体为自然群体,不需要耗费大量时间构建双亲作图群体,极大地节约了时间和降低了工作强度;2)可同时鉴定多个等位基因。全基因组测序技术的不断发展推动了全基因组关联分析(genome-wide association study,GWAS)技术用于遗传作图的进程。GWAS 又名连锁不平衡作图,可快速对QTL 进行精细定位,且分析通量较高[8]。该方法在研究水稻的农艺性状改良中被广泛应用。如Lu 等[9]利用469 份水稻种质资源定位了17 个水稻剑叶长度相关的QTL;Huang等[10]从950 份水稻品种中定位到10 个与产量性状相关的QTL;Jiang 等[11]鉴定出350 份水稻品种中23 个与水稻分蘖相关的QTL。近几年GWAS 也被应用于定位抗病相关位点。如Zhang 等[12]用6 个白叶枯小种对172 份群体材料进行接种,通过GWAS鉴定了12 个白叶枯抗性相关位点;Kang 等[13]用5 个稻瘟菌接种了390 份群体材料,鉴定出97 个稻瘟病抗性相关位点;Liu 等[14]通过GWAS 鉴定了27 个稻瘟病抗性相关位点,并成功克隆了一个稻瘟病抗性基因PiPR1。GWAS 方法的广泛应用极大程度上提高了育种的效率及精度,为后续抗性品种选育提供了一定的科学基础。
为进一步发掘新的稻瘟病抗性基因,本研究对水稻多样性群体II(rice diversity group II,RDP-II)中470 份水稻种质资源进行了稻瘟病抗性评估,并使用高密度阵列[700 000 个单核苷酸多态性(single nucleotide polymorphism,SNP)]进行了基因分型[15],选择最优的关联分析模型,并对定位的稻瘟病抗性相关的QTL 进行了分析,为快速发掘新的稻瘟病抗性基因提供了科学依据。
1 材料与方法
1.1 试验材料与田间种植
本研究所用的470 份水稻材料来自RDP-II,由广东省农业科学院刘斌老师提供。于2018 年5 月将育好的秧苗种植于湖南省益阳市桃江县的自然病圃中(N28°22',E112°03'),以每行20 株种植24 行,株行距为15 cm×24 cm。该病圃面积约0.133 km2,历年来稻瘟病发生较为严重。此外,为保证发病质量,本研究在群体材料周围种植了高感稻瘟病的品种进行自然诱发。在生长阶段应注意水培管理,多施加氮肥以诱导稻瘟病的发生,待水稻苗三叶一心时期(2018年6月)进行叶瘟调查。
1.2 表型调查及分析
根据稻瘟病分级标准对自然病圃中群体材料进行调查[13],其中0 级(高抗)为无病斑;1 级(高抗)为仅有针尖大小病斑;2 级(抗)为出现较大褐点;3 级(中抗)为直径1~2 mm 的小圆斑;4~6 级(中感)开始形成典型的病斑,占叶面积大小约6%~25%;7 级(感)的病斑面积超过25%;8~9 级(高感)的病斑面积达到50%以上,甚至整片叶片死亡。利用Excel 2010 进行表型数据的处理,过滤掉不正常数据后,以2 次重复数据的平均值对亚群表型及抗性品种分布等结果进行分析。
1.3 基因型检测与分析
利用Mccouch 等[15]报道的700 000 个SNP 芯片对470 份水稻种质资源进行基因型分析,在Tassel 4.0 软件中过滤掉低质量的SNP 位点,选择最小等位基因频率(minor allele frequency,MAF)>0.05 以及缺失率≥20%的SNP 位点,以进行下一步的关联分析。
1.4 GWAS 数据分析
本研究利用700 000 个SNP 基因型数据以及来自RDP-II 群体的470 份水稻材料的抗病表型数据,选择混合线性模型(mixed linear model,MLM),即同时加入亲缘关系Kinship 以及群体结构Q 矩阵,在Tassel 4.0软件中进行GWAS。利用R软件(R version 3.6.2)的qqman包绘制曼哈顿图。
1.5 抗病相关QTL的定位
通过GWAS,我们得到176 个与稻瘟病抗性相关联的SNP[-log10(Pvalue)≥3.50]。为了降低假阳性,我们以在200 kb 的基因组间隔内具有至少2 个显著相关的SNP 位点为标准定位抗病相关的QTL。在国家水稻数据中心(http://www.ricedata.cn/)及NCBI 数据库(https://www.ncbi.nlm.nih.gov/)中寻找已克隆或定位的稻瘟病抗性相关基因的位置信息,与本研究所检测的QTL进行共定位。
2 结果与分析
2.1 自然群体抗病表型分析
本研究根据国际水稻所稻瘟病0~9 级分级标准,对湖南省桃江县自然病圃中(图1)的470 份水稻品种进行了稻瘟病抗性的评估,其中0~1 级为高抗、2 级为抗、3 级为中抗、4~6 级为中感、7 级为感、8~9级为高感。
抗病表型的统计结果如表1 所示。抗病品种占自然群体的32.98%,而感病品种占自然群体67.02%,表明该群体总体趋势上对稻瘟菌表现为感病。

图1 湖南省桃江县稻瘟病自然病圃Fig. 1 Natural blast nursery in Taojiang county, Hunan province
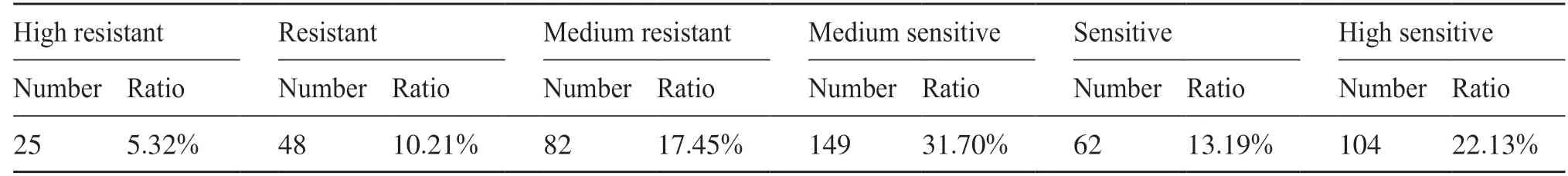
表1 RDP-II群体中抗病表型分布Tab. 1 The distribution of resistance phenotype in RDP-II population
2.2 群体中亚群抗病表型分析
本研究所用群体材料除6 份未定义品种外,可大致分为6 个亚群,其中籼稻(indica,IND)163 份、香型稻(aromatic,ARO)65份、热带粳稻(tropical ja-ponica,TRJ)102份、温带粳稻(temperate japonica,TEJ)35份、混合型水稻(admixture,ADM)28份,奥斯稻(aus,AUS)71 份。从亚群抗病表型直方图中(图2a)我们发现,热带粳稻亚群的平均抗性水平最高,达到4.0,而温带粳稻亚群的平均抗性水平最低,达到6.0,籼稻亚群的平均抗性水平居中,为5.3。气泡图如图2b 所示。气泡自上而下分别代表1~9 抗病等级,气泡大小代表品种数目。通过分析发现,在抗病等级≤3(表型表现为抗病)时,热带粳稻亚群中含有56个符合条件的品种,而温带粳稻亚群仅含有8个品种。该结果进一步证明了以上结论。
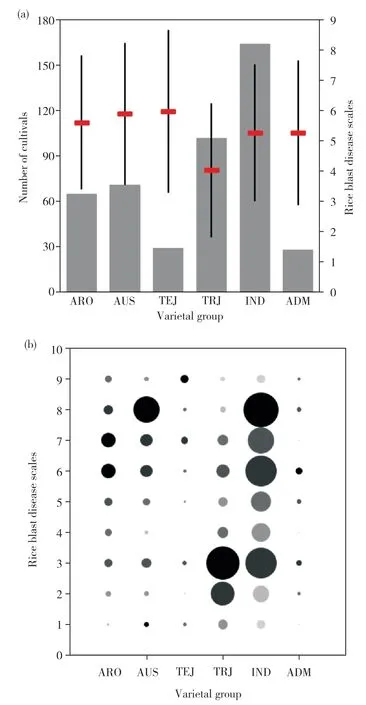
图2 RDP-II群体的亚群抗病表型Fig. 2 Subpopulations resistance phenotype of RDP-II
通过对470 份水稻群体材料的表型数据进行统计发现,在73份抗性品种(0~2级)中,热带粳稻亚群的品种最多,占总数的38%,而温带粳稻亚群的品种仅占6%。该结果同群体材料中亚群稻瘟病抗性分布较为一致。此外,表2 特别列出了超高抗的25 个品种(0~1 级),其分别来自亚洲(56%)、非洲(24%)、北美洲(16%)以及欧洲(4%)这4个不同的大洲。这些超高抗材料为抗病育种提供了优良的种质资源。
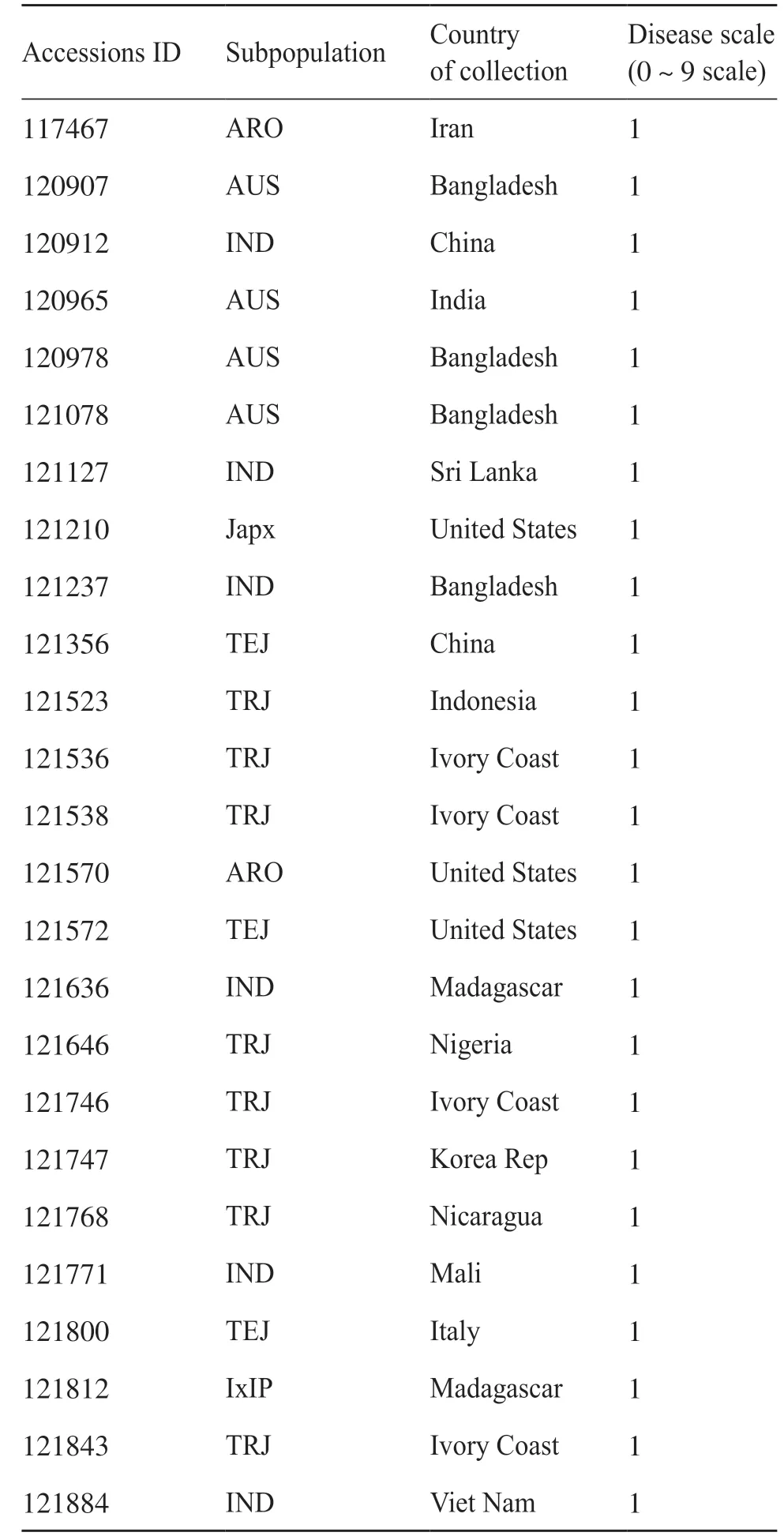
表2 高抗品种亚群及地区分布Tab. 2 Subpopulation and regional distribution of high resistant varieties
2.3 GWAS
本研究基于470 份水稻自然群体材料的抗病表型数据以及700 000 个SNP 基因型数据, 选择MLM模型进行抗病表型数据和SNP 标记的GWAS。在阈值[-log10(Pvalue)≥3.50]时判定SNP 标记与抗病表型关联的显著性,共检测到176 个相关联的SNP位点,覆盖于全部染色体。抗病表型关联分析结果如图3 所示,曼哈顿图中红色虚线以上代表抗病相关性显著的位点。
2.4 稻瘟病抗性关联位点鉴定
以SNP 上下游200 kb 范围内至少2 个显著相关SNP 位点作为1 个QTL,对GWAS 结果所检测的176 个抗病相关SNP 进行QTL 分析。由表3 可知,在除了4 号及7 号外的10 条染色体上定位到24 个稻瘟病抗性相关位点(loci associated with rice blast resistance,LABRs)。6 号染色体上分布最多,为4 个,其中最关联的SNP 标记(SNP-6.8792603)也位于6 号染色体上。将这些检测的LABRs 同已克隆或定位的稻瘟病抗性相关基因进行共定位,结果共定位到4 个已克隆稻瘟病抗性基因和2 个已定位的基因,分别是1 号染色体的OsSGT1[16]、2 号染色体的OsCBSX3[17]和OsCUL3a[18]、11 号染色体的Pi-38[19]、12 号染色体的Pi-ta[20]和Pi-42[21]。分析发现,这4 个基因中除了OsCUL3a为负调控因子外,其余均为抗病反应的正调控因子。
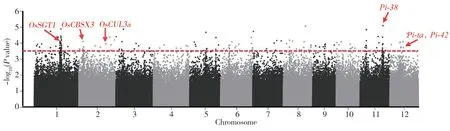
图3 稻瘟病抗病表型全基因组关联分析曼哈顿图Fig. 3 Genome-wide association analysis manhattan plots of rice blast resistance
3 讨论
稻瘟病抗性机制复杂,既受主效基因的控制,又受QTL 的影响。由主效基因介导的抗性是定性的,对稻瘟菌具有完全的抗性,而由多个QTL 介导的抗性是定量的,对稻瘟菌只表现部分抗性[22]。广泛的实践证明,单一主效抗性基因在3~5年内易失去抗性[23],因此快速挖掘新的稻瘟病抗性基因意义重大。本研究对470 份水稻种质资源进行了水稻苗期稻瘟病抗性评估,其中热带粳稻亚群的平均抗性水平最高,而温带粳稻亚群的平均抗性水平最低,籼稻亚群的平均抗性水平居中。该结论与之前对RDP-I 和RDP-II 群体的表型研究结果一致[13-14]。同时对高抗品种的亚群分布进行研究,发现来自热带粳稻亚群最多,这与群体分布结果相一致。此外,高抗品种中一半以上来自亚洲(56%),可能与亚洲地区的稻瘟病抗性选择较高有关。本研究所获得的高抗品种为育种家提供了新的抗稻瘟病种质资源。
本研究选择混合线性模型MLM 对抗病表型数据及700 000 个SNP 基因型数据进行GWAS,共检测到分布于12 条染色体上的176 个相关联SNP位点。通过对这些SNP 位点进行QTL 分析发现,在除了4 号及7 号外的10 条染色体上定位到24 个LABRs,-log10(Pvalue)值介于3.72~5.10 之间,可以解释3.37%~45.06%的表型变异。将这些LABRs同已克隆或定位的基因进行共定位,分析发现,5 个位点(LABR2、LABR4、LABR6、LABR22、LABR23)共定位了6 个稻瘟病抗性相关基因,包括4 个已克隆及2 个已定位的基因,分别位于1、2、11 及12 号染色体上。这5 个位点对应的峰值SNP 分别为SNP-1.25004091、SNP-2.3910977、SNP-2.31170935、SNP-11.21704502、SNP-12.10723430。除上述5 个位点与已报道的位点共定位外,其余19 个显著关联位点为首次发现。这些新位点的发现对后续稻瘟病抗性基因的发掘以及进一步的优良品种选育工作具有重要意义。基于以上研究结果,下一步的研究工作将主要集中于候选基因的选择以及稻瘟病抗性基因的克隆与功能验证,为培育广谱抗性水稻品种提供理论基础。
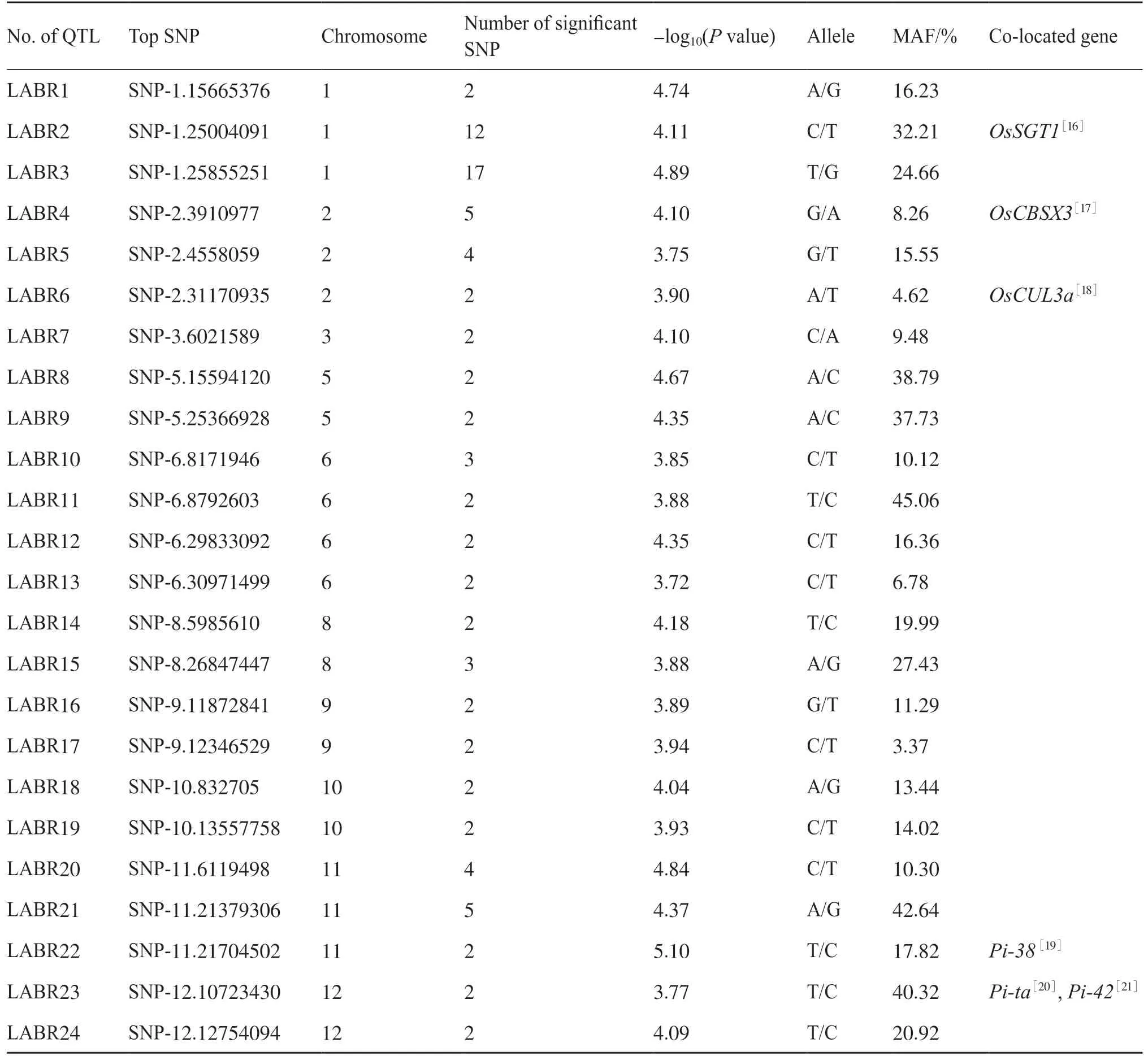
表3 24个稻瘟病抗性显著相关位点Tab. 3 24 significant LABRs
猜你喜欢
现代临床医学(2023年1期)2023-03-24
今日农业(2022年4期)2022-06-01
中国医药科学(2022年5期)2022-05-05
作物学报(2022年6期)2022-04-08
浙江农业学报(2017年1期)2017-05-17
河南农业(2016年6期)2016-11-26
新农业(2016年20期)2016-08-16
现代农业(2016年5期)2016-02-28
中国卫生标准管理(2015年3期)2016-01-14
现代农业(2015年1期)2015-02-28
