
猪肉中常见的9种蓄水类药物残留的快速筛查
2021-05-14 00:30陈树兵余晓玲方科益余晓琴王传现
分析科学学报 2021年2期
陈树兵,余晓玲,李 双,方科益,余晓琴,韩 超,王传现
(1.宁波海关技术中心,浙江宁波 315040;2.四川省食品药品检验检测院,四川成都,610097;3.浙江树人大学生物与环境工程学院,浙江杭州 310015;4.上海海关动植物与食品检验检疫技术中心,上海 200135)
我国作为猪肉消费大国,其肉制品的生产安全直接关系到民生建设。近些年,出现的“注水肉”问题与非法使用新型蓄水类药物直接相关。新型蓄水类药物主要包括:阻断M胆碱受体的抗胆碱药物(阿托品、东莨菪碱、山莨菪碱),局部麻醉药物(普鲁卡因、利多卡因),抗组胺类药物(异丙嗪)和肾上腺素受体激动药物(肾上腺素及其代谢物4-羟基-3-甲氧基-扁桃酸和3,4-二羟基扁桃酸)[1 - 10]。这些药物的非法使用将不可避免地残留于猪肉食品中,进而对人体中枢神经造成不良影响。目前,我国蓄水类药物残留检测标准缺乏,已有文献多是按照某一类药物建立的,适应范围单一[1,2,8,11,12],尚无同时测定猪肉中9种蓄水类药物及其代谢物的报道。因此,建立猪肉中蓄水类药物的高通量筛查方法,对于提高监管工作效率、保障猪肉食品安全具有重要的意义。
蓄水类药物检测主要涉及前处理技术和仪器分析两方面。已有方法主要采用固相萃取柱净化。例如:王海燕等[1]对畜肉中阿托品类药物残留量测定,前处理采用MCX小柱净化;胡海山等[8]对动物源性食品中镇静剂类药物残留量测定,乙腈提取后经Oasis HLB小柱净化;另外,段科等[5]对肉中的阿托品类药物残留量测定,乙腈提取后正己烷除油净化。采用固相萃取柱净化,步骤复杂且实验成本较高,而乙腈作为提取液,没有兼容到强极性物质的提取,对偏极性的蓄水类药物不能保证回收率。如何把极性物质从水溶液中有效提取出来,这始终是一个艰巨挑战。随着质谱的发展和推广,一次进样完成多类药物残留分析已成为可能。药物残留正逐渐由液相色谱-三重四极杆质谱(LC-MS/MS)目标型检测向高分辨质谱(HRMS)非目标型全扫描检测转变。与传统的LC-MS/MS法相比较,高分辨质谱仪可直接采集高准确度质量数(m/z200,分辨率70 000),大大降低了近似质量数的干扰,通过自动触发二级,避免了假阳性结果的发生,特别适合高通量的筛查检测。本文利用载体辅助液-液萃取技术,通过一次前处理实现了9种常见注水类药物的提取净化,同时结合四极杆静电场轨道阱高分辨质谱检测,为提高监管工作效率,保证动物源性食品安全提供了基础。
1 实验部分
1.1 仪器与试剂
Q-Exactive四极杆静电场轨道阱高分辨质谱仪(赛默飞世尔科技公司),配有H-ESI Ⅱ源。UltiMate 3000液相色谱系统,配有自动进样器。硅藻土柱型号为Chromabond XTR(1 000 mg)。
9种蓄水类药物标准品购自Sigma和Dr.Ehrenstorfer公司,纯度≥95%。二甲基亚砜(分析纯)购自南京化学试剂一厂。提取液的配制:称取4.3 g的H2C2O4,用500 mL水溶解,氨水调节pH至3.0。色谱纯甲酸购自美国Sigma-Aldrich公司。其他试剂均为色谱纯,购自德国Merck公司。实验用水为Milli-Q超纯水(18.2 ΩM·cm)。
1.2 提取净化流程
基质样品来自国家猪肉残留监控抽样和进出口送检企业。称取5.00 g样品,加入10 mL提取液,振荡、各超声5 min,4 500 r/min下离心5 min;取水相上大孔硅藻土柱后,平衡5 min,用10 mL乙腈洗脱2次,下接50 mL离心管,并加入0.5 mL二甲基亚砜,乙腈定容至20 mL;取出10 mL,氮吹后用纯水定容至1.0 mL,过0.22 μm 滤膜,待分析。
1.3 色谱及质谱条件
色谱柱:Hypersil Gold C18柱(100 mm×2.1 mm,1.9 μm);流动相A:0.1%甲酸水溶液;流动相B:乙腈。梯度洗脱:0~3 min,B相保持5%;3~6 min,B相5%~90%;6~8 min,B相保持90%;8~8.1 min,B相90%~5%。流速0.3 mL/min;柱温40 ℃;进样量10 μL。
质谱条件:质谱在正/负离子转换模式下进行全扫描,质量范围:m/z100~500,分辨率70 000,自动增益控制(AGC)目标值5.0×105;4-羟基-3-甲氧基-扁桃酸和3,4-二羟基扁桃酸采用负离子模式2 700 V,其余成分则采用正离子模式3 800 V;离子传输管温度为300 ℃;鞘气流速(N2)35 L/h;辅助流速(N2)10 L/h;气化室温度350 ℃。在样品运行前对仪器分别进行正、负离子校正;二级采用自动触发模式,分辨率35 000,AGC目标值2.0×105,碰撞能量范围25%~40%,保留时间采集范围:根据一级色谱图中各个目标物的保留时间值±1.0 min。
1.4 定性、定量分析
定性分析时精确质量误差低于5.0×10-6,同时比对保留时间、同位素分布、主要二级碎片和二级质谱图相似度,综合判断,避免假阳性结果出现。
空白样品按照“1.2”处理,在得到的基质溶液中加入上述9种目标物混合标准溶液,配制基质加标溶液并进行测定,以浓度为横坐标,仪器响应值(响应值=标准品峰面积/标准品质量)为纵坐标做标准曲线,作为样品中待测物浓度定量的依据。其中,高浓度加标样品应稀释至标准曲线范围内进行定量分析。
2 结果与讨论
2.1 提取液的选择
考虑到猪肉基质的高脂肪属性,且9种待测物均为碱性化合物,水溶性强,方法采用酸化的水溶液进行提取,在保证9种蓄水类化合物高提取率的同时,又可以防止脂肪被过多的提取出来,降低脂肪的干扰。H2C2O4含有的两个羧基基团,较其他可挥发性甲酸、乙酸,具有更强的稳定性和酸性,此外H2C2O4作为金属螯合剂,可大大降低金属离子的干扰。
2.2 提取剂的添加量对柱填料的影响
提取液的添加量要充分考虑柱子填料的含量,本文选择硅藻土柱Chromabond XTR(1 000 mg)。实验中分别比较了上样量为5 mL、8 mL、10 mL、12 mL、15 mL时柱子的负载情况,在上样量达到12 mL和15 mL时,柱子已达到饱和并渗出,水溶液的渗出会导致氮吹浓缩过程大大延长。另外,当上样量为5 mL和8 mL时,柱子的填料未充分浸入水溶液,大大降低了柱子的利用率,同时对于9种待测药物本就很低的检出限量来说,也增加了仪器检出的难度。因此,本研究最终将上样体积确定为10 mL。

图1 乙腈洗脱体积对9种注水类药物回收率的影响Fig.1 Effect of elution volume of acetonitrile on recovery of 9 waterflooding drugs
2.3 洗脱体积的优化
水相提取液上样后,以纯乙腈作为洗脱液,通过乙腈的逐级渗入,注水类药物按照极性强度依次从饱和的硅藻土柱上洗脱下来。实验中分别考察乙腈作为洗脱溶剂的添加量为10 mL、10 mL×2和10 mL×3对待测物回收率的影响。由图1可见,当乙腈的体积为10 mL×2和10 mL×3时,提取率趋于稳定,9种待测注水类药物在猪肉中的回收率均保持在80%以上。从节约氮吹浓缩的时间和成本的角度出发,确定纯乙腈作为洗脱液的洗脱体积为10 mL×2。
2.4 仪器条件优化
通过注射泵连续进样,对上述蓄水类药物的单标准溶液进行分析,获得每种化合物的最佳电离方式和分子离子峰的存在形式。其中4-羟基-3-甲氧基-扁桃酸和3,4-二羟基扁桃酸采用ESI-模式,其余化合物均采用ESI+模式。
流动相中加入0.1%甲酸能增加正离子模式检测下物质的电离效率,促进[M+H]+离子生成,同时进一步考察NH4Ac添加量对待测化合物的影响,NH4Ac从2 mmol/L到10 mmol/L,结果表明9种蓄水类药物在0.1%甲酸溶液中均获得了最佳的色谱峰形、分离效果和质谱信号响应。从减小仪器污染的角度出发,本文最终采用含有0.1%甲酸水溶液作为流动相A,全程采用梯度洗脱,实现了9种待测物的有效分离。9种注水类药物的提取离子色谱图见图2。

图2 9种蓄水药物的提取离子色谱图Fig.2 Extraction ion chromatograms of 9 waterflooding drugs
2.5 加标回收结果分析
结果表明,肾上腺素及其代谢物4-羟基-3-甲氧基-扁桃酸在5.0~50.0 ng/mL浓度范围内,其他药物(异丙嗪,阿托品、山莨菪碱、东莨菪碱、普鲁卡因和利多卡因)在0.5~5.0 ng/mL浓度范围内,各自呈现良好的线性关系,相关系数r2均大于0.990,满足定量要求。9种待测物的回收率在80.0%~95.2%之间,相对标准偏差低于10%。用基质提取液添加低水平的标准溶液(肾上腺素及代谢物为5.0 μg/kg,其他待测物为0.5 μg/kg),获得每种待测物对应信噪比(S/N)≥10时的含量,以此为该化合物的定量限(LOQ)(表1)。结果表明,该方法灵敏度高,操作简便,适用于猪肉中蓄水类药物残留的快速筛查。
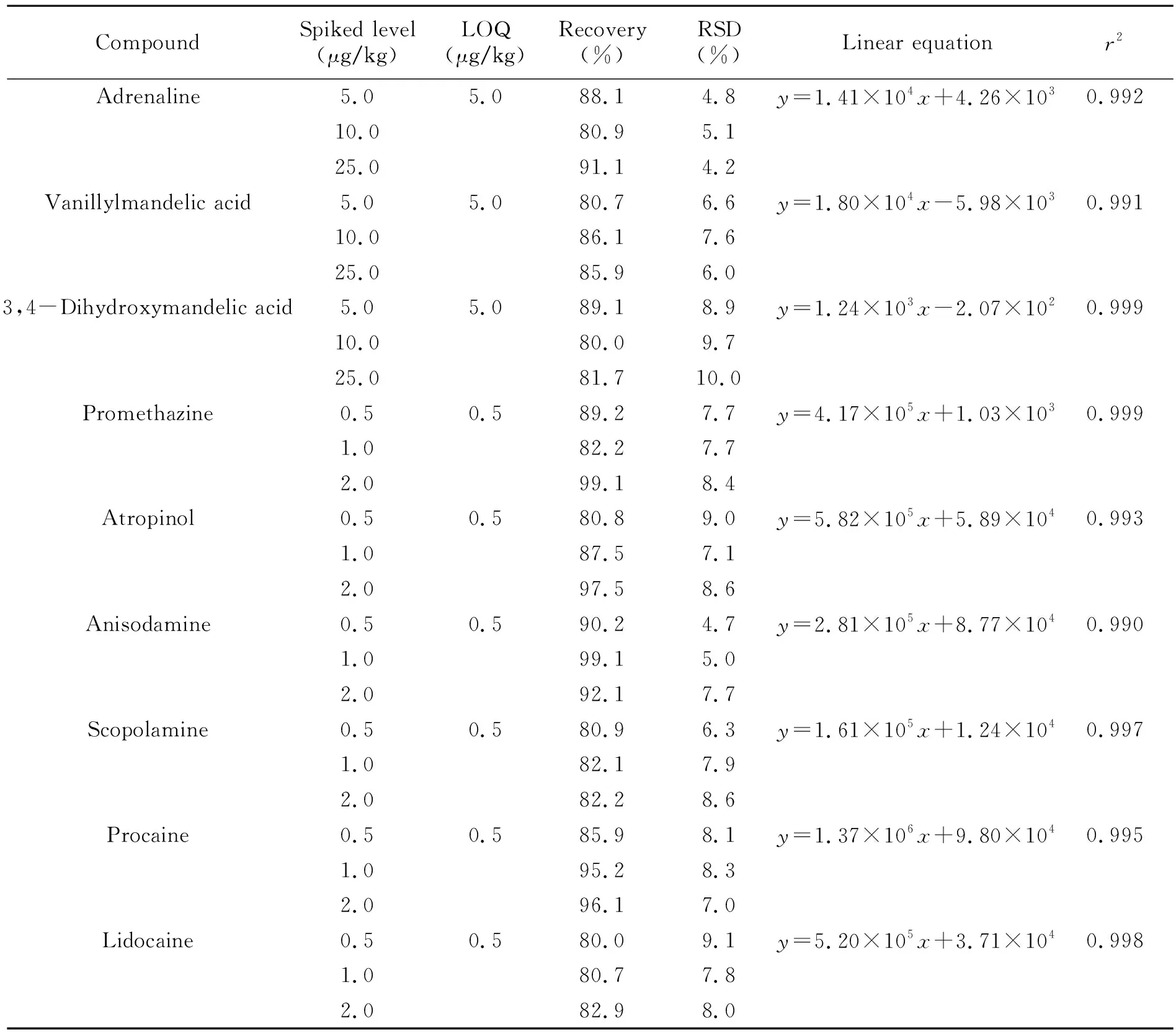
表1 猪肉中9种注水类药物的定量限(LOQs)、线性方程及不同添加浓度的平均回收率(n=6)
3 结论
本研究建立了猪肉中常见的9种蓄水类药物:肾上腺素及其代谢物4-羟基-3-甲氧基-扁桃酸和3,4-二羟基扁桃酸、异丙嗪、阿托品、山莨菪碱、东莨菪碱、普鲁卡因和利多卡因的快速筛查方法。待测物经水溶液提取后,利用硅藻土柱完成水溶性药物的同时提取、净化、浓缩过程。同时结合四极杆静电场轨道阱高分辨质谱,实现了上述药物残留的“一步式”多残留筛查。该方法操作简单,重现性好,定量限低,完全满足国标中规定的猪肉中9种蓄水类药物的测定需求。
猜你喜欢
中草药(2022年19期)2022-10-14
农产品加工(2022年4期)2022-03-11
商品与质量(2021年43期)2022-01-18
建材发展导向(2021年7期)2021-07-16
昆明医科大学学报(2021年1期)2021-02-07
中国科技教育(2017年9期)2018-01-23
食品界(2017年12期)2018-01-20
能源研究与信息(2014年3期)2014-10-30
东方艺术·大家(2014年4期)2014-07-18
数理化学习·高一二版(2009年1期)2009-03-19
