
吹扫捕集-原子荧光光谱与同位素稀释质谱法结合测定海水中痕量汞
2021-05-14 00:30巢静波王静如李芳缘
分析科学学报 2021年2期
巢静波,王 茜,王静如,李芳缘,陈 艳
(1.中国计量科学研究院化学计量与分析科学研究所,北京100029;2.北京化工大学化学工程学院,北京100029)
由于自然和人类的活动,汞早已成为一种全球性的环境污染物[1]。海洋作为大气汞的源和汇,在全球汞的循环中起着主导作用[2]。在海洋环境中,汞经生物甲基化作用可转化成毒性更强的甲基汞,研究表明食肉鱼类中甲基汞浓度可达海水中汞浓度的1~10万倍[3,4],鉴于其在环境和生物中的累积性和持久性[5],汞已成为对海洋生物毒性最强的元素之一,也因此成为水环境的重要监测目标物。我国国家标准(GB 3097-1997)《海水水质标准》规定四类海水中汞的限量值分别为0.05 μg/L、0.2 μg/L、0.2 μg/L和0.5 μg/L[6],在此痕量和超痕量水平上,很容易在样品处理和分析过程中造成污染、挥发或者吸附损失[7],进一步加大了准确检测的难度。有文献报道海水中汞的浓度约为1.0×10-11mol/L(2 ng/L)[8],然而后续研究表明:在样品采集、处理和分析过程中的污染严重损害了很多已报道结果的可靠性,在某些情况下,汞的测量值甚至是其真实浓度的100倍[9,10],这与天然水中总汞的真实浓度约在0.2~100 ng/L的估计值[11]基本一致。海水中除溶解的Hg2+外,还含有少量甲基汞等有机汞形态。另外,汞极易与有机分子和碳形成稳定共价键[12,13],因此极易吸附在海水中的颗粒物以及保存容器上。基于上述原因,海水中汞的测定浓度可能在很大范围内变化[14 - 16],这给环境监测、污染调查、相关政策制定等工作带来很大不确定性。因此,发展高灵敏度、高准确性的检测方法成为获得真实可靠结果的前提和关键。
目前,环境水体中汞的测定方法主要有冷原子吸收法[1,12,17,18]、冷原子荧光法[19 - 22]、电感耦合等离子体质谱法(ICP-MS)[7,23]、电化学法[24,25]等。上述三种原子光谱法较电化学法具有较高的灵敏度,但仍难以直接检测海水中的痕量、超痕量汞。除海水中汞的含量极低、在测定过程中容易引入外源污染是主要原因外[24],另一个难点是海水的高盐基体(约35 g/L)导致直接和间接的光谱干扰,且在ICP-MS测定时需要进行10倍以上稀释,或者采用稀释气体以防止采样锥的堵塞,另外在测定过程中汞极易吸附在进样系统中而产生记忆效应。美国EPA 1631是目前被广泛应用和认可的痕量汞测定方法,该方法是利用吹扫-金汞齐捕集的方式,实现汞与海水基体的分离,采用原子荧光光谱法实现汞的高灵敏度检测。
本研究结合吹扫-金汞齐捕集前处理方法,利用原子荧光直观观测和同位素稀释质谱法高准确度的特点,首先采用吹扫-金汞齐捕集-原子荧光总汞测定仪离线观测仪器本底,并考察和优化盐度、氧化用HNO3浓度和氧化时间、还原剂用量、本底控制等条件;待本底稳定后直接将总汞测定仪与ICP-MS联用,以同位素稀释质谱法进行海水中痕量汞的高准确度测定。建立的方法应用于渤海、黄海海水和国际比对海水样品中痕量汞的测定,并对测定结果进行了不确定评定,以评估该方法的性能参数和计量学特性。
1 实验部分
1.1 主要仪器与装置
8800型三重四极杆电感耦合等离子体质谱仪(美国,Agilent公司),配有数据采集及处理软件(MassHunter Workstations);MERX全自动总汞分析系统(美国,Brooks Rand Labs公司);XP205型电子分析天平(瑞士,Mettle -Toledo公司);Integral 5型超纯水机处理系统(美国,MilliQ公司);FP-31型马弗炉(日本,YAMATO公司);磁力搅拌器(德国,IKA公司)。
MERX全自动总汞分析系统与ICP-MS联用时,是将经SnCl2还原后的样品通过自动进样器加入到气-液分离瓶中,还原后形成的汞被高纯氮气吹出,进入金柱进行金汞齐捕集,随后进入热脱附装置中进行热脱附,由载气带入原子荧光检测器,最后通过聚四氟乙烯三通与ICP-MS载气混合后,经矩管连接支管(前端用封口膜进行封口)引入ICP-MS系统进行检测。为保持样品进样与ICP-MS检测的同步,通过设计的信号线在样品热脱附裂解时触发ICP-MS数据采集信号,实现样品的自动测定。
1.2 主要试剂与材料
GBW(E)080124汞单元素溶液标准物质(100 μg/mL),相对扩展不确定度为0.8%(k=2),购自中国计量科学研究院;GBW04443202Hg浓缩同位素稀释剂标准物质购自中国计量科学研究院,浓度标准值为2.978×10-8mol/g,扩展不确定度为2.4×10-10mol/g(k=2);200Hg/202Hg同位素丰度比标准值为0.003387,扩展不确定度为0.000086(k=2);SnCl2·2H2O为优级纯,购自天津市光复精细化工研究所;KBr、KBrO3、盐酸羟胺均为分析纯,购于国药集团化学试剂有限公司;HNO3(TAMAPURE-AA-10)购自日本多摩化学工业株式会社;HCl为优级纯,购自北京化学试剂研究所;20%SnCl2溶液:称取20 g SnCl2·2H2O溶于10 mL热HCl,完全溶解后用水稀释至100 mL;30%盐酸羟胺:称取15 g盐酸羟胺溶于50 mL水中;BrCl溶液:将2.7 g KBr溶于250 mL HCl中,磁力搅拌1 h后,再缓慢加入3.8 g KBrO3,再搅拌1 h。Li、Y、Ce、Tl、Co质谱调谐溶液(1 ng/mL)由Agilent公司提供。实验样品瓶为40 mL棕色玻璃瓶(Brooks Rand Labs公司),带有硅胶垫。
3种海水样品分别采自马尔马拉海(Marmara Sea,E27,N40)、渤海(E120,N39)、舟山岛附近(E122,N30)表层海水,其中马尔马拉海水为“CCQM-K155&P196海水中元素和三丁基锡”国际比对样品,该比对样品采集后用重蒸HNO3调节pH为1.5并放置2周后,采用0.22 μm滤膜过滤,混匀后分装至经HNO3、稀金溶液和水反复洗涤的低密度聚乙烯塑料瓶中,每瓶250 mL。渤海海水和舟山岛海水分别为海水中金属元素和营养盐标准物质候选物,前者采集后用1%HNO3酸化并于低密度聚乙烯塑料桶陈化半年;后者未经酸处理,于低密度聚乙烯塑料桶放置1年,测定前采用0.22 μm滤膜过滤。
1.3 样品测定
为降低仪器本底,测定前向样品瓶中加入25 mL水和50 μL 20%SnCl2溶液,对所有样品瓶进行反复吹扫和金捕集柱加热脱附,以离线方式用原子荧光检测器监测本底信号,待信号稳定后进行样品测定。
准确称取海水样品2.0~3.0 g(精确到0.00001 g)于40 mL棕色样品瓶中,分别向三种海水样品中加入准确称重的0.1~0.2 g约为0.3 ng/g的202Hg同位素稀释剂,混匀。加入100 μL HNO3,静置5 h或者60 ℃加热30 min后,加入水至25 mL,再加入150 μL 20%SnCl2溶液,盖紧瓶盖,置于自动进样器待测。同时做空白实验。样品测定前,以水相进样的方式离线调节ICP-MS进样和检测系统参数,优化灵敏度的同时使氧化物和双电荷比率分别小于1%和3%。优化完成后重新连接总汞分析系统载气和ICP-MS稀释气管路,开机预热30 min后开始检测,以峰面积进行定量。
总汞分析系统工作参数:吹扫捕集气流速为350 mL/min,载气流速为30 mL/min,吹扫时间为6 min;捕集管加热、干燥和冷却时间分别为2.5 min、3 min和1 min。
ICP-MS仪器工作参数:RF功率为1 550 W,采样深度为9.5 mm,载气流速为0.95 L/min,数据采集模式为时间分辨(TRA),积分时间为0.3 s。基于金柱捕集、脱附、干燥和冷却时间,ICP-MS设置采集时间为6 min,汞的出峰时间为50 s,监测比值为200Hg/202Hg。
2 结果与讨论
2.1 本底控制及记忆效应
如前所述,为保证测定结果的精密度和准确度,防止在样品前处理和测定过程中汞的沾污,并进一步降低本底和记忆效应,首先将棕色样品瓶去盖后置于马弗炉中,于500 ℃条件下空烧30 min;同时对配制的20%SnCl2溶液用氮气缓慢吹扫0.5 h,去除还原剂中的汞。测定样品前向样品瓶中加入适量SnCl2溶液进行反复吹扫,并对金捕集柱加热脱附,用原子荧光检测器监测本底信号。测试结果表明,经过反复吹扫后本底可降至3.5 pg。根据测定信号选择本底水平一致的样品瓶用于后续实验,且在每次实验前重复上述操作以控制本底。对于ICP-MS检测系统,由于样品是以气体方式进样,汞在进样系统中的记忆效应本身很小。总汞分析系统包括两根金捕集柱,以交替方式进行汞的捕集。当高含量样品进样后,在相应捕集柱对应的样品位置放置清洗样品,进行再次脱附后可较好地消除记忆效应。
2.2 SnCl2的加入量
为考察SnCl2的还原效果,分别向1 ng Hg标准中加入20、50、100、150和200 μL氮吹处理过的20%SnCl2溶液,并同时做空白扣除,以原子荧光光谱获得的峰面积对结果进行比较。从图1中可以看出,当SnCl2溶液加入量为150 μL时,获得的信号值最大,故后续的实验选择其加入量为150 μL。
2.3 基体的影响
在进行原子荧光测定时,汞通过还原成气态实现与海水基体的分离,但高盐含量可能会影响单位时间内进入捕集系统的汞总量,在进行外标法测定时,需要考虑标准曲线与海水样品进行基体匹配,另外高盐的存在会影响碱石灰的脱水效果。近岸海水中NaCl的含量一般在2%~3%之间,为考察海水基体对汞测定的影响,分别向1 ng Hg标准中加入0%、0.5%、1.0%、1.5%、2.0%、2.5%和3.0%的高纯NaCl,同时做空白扣除,测定结果见图2。从结果可以看出,随着NaCl含量增加,Hg的含量测定值明显增高,这可能是由于盐效应导致相同吹扫时间内进入金捕集柱Hg含量增加导致的。当NaCl含量为0.5%时,测定结果较不加NaCl样品高约2.2%,因此在进行原子荧光测定时,海水的取样量应控制在盐含量小于0.5%。同位素稀释质谱法为潜在基准测定方法,在稀释剂与样品混合均匀的情况下,测定结果不受样品损失以及金捕集柱吸附、脱附效率影响,但考虑到高盐可能会造成整个富集系统吸附、脱附效率降低,从而导致灵敏度下降,因此后续实验样品取样量控制在2~3 g,并采用同位素稀释质谱法对样品进行准确测定。

图1 SnCl2加入量的影响Fig.1 Effect of SnCl2 volume on Hg detection
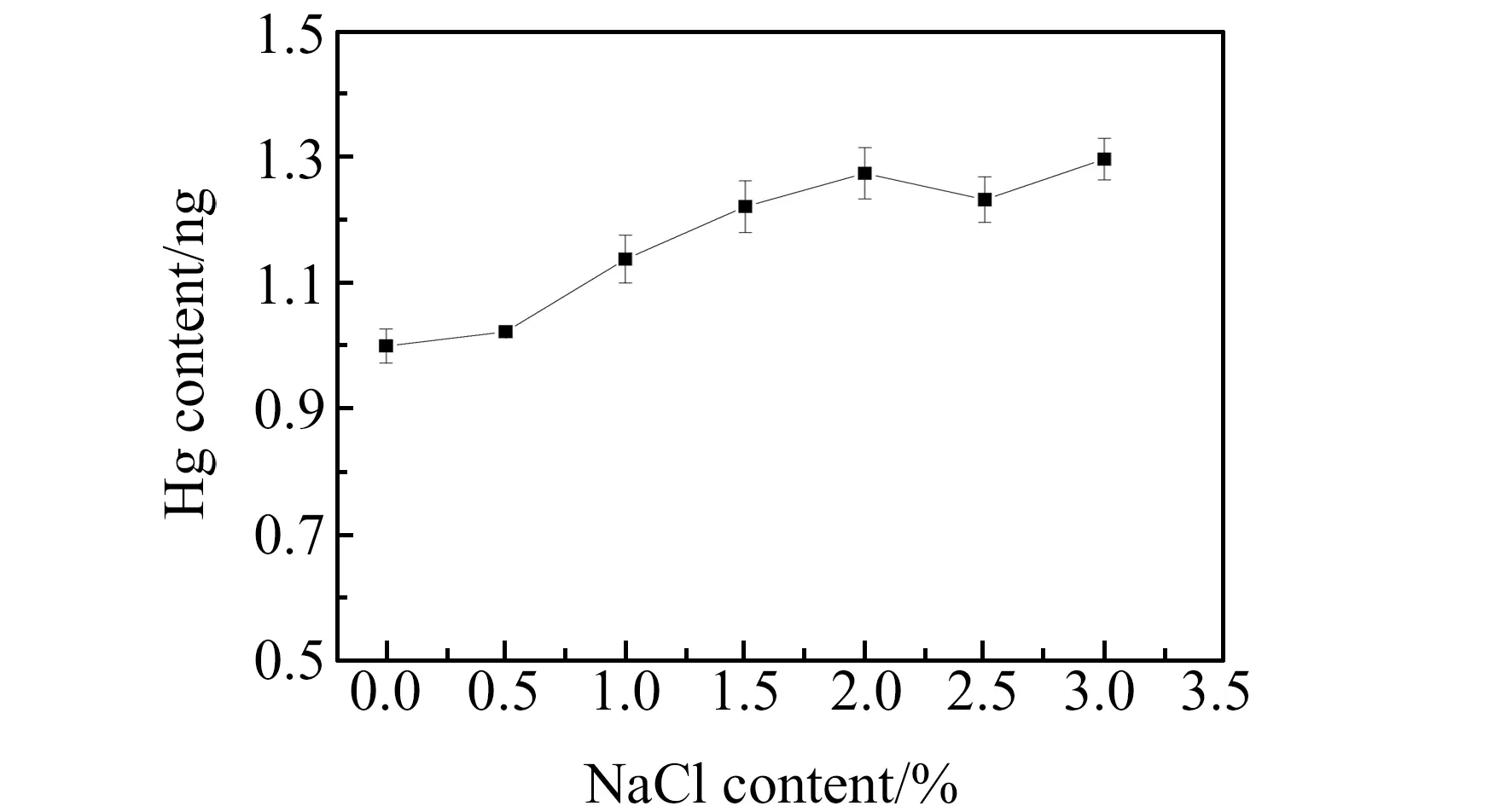
图2 NaCl含量对测定结果的影响Fig.2 Effect of NaCl content on Hg detection
2.4 HNO3加入量的优化
经测定,3个海水样品中NaCl含量约为2.0%~2.5%,由于海水样品均经过陈化和过滤,因此与颗粒物结合的汞含量较低,为避免由于多种试剂加入引起汞的本底升高且不易控制,本研究采用HNO3作为氧化剂。以国际比对海水样品考察HNO3加入量对测定结果的影响。向样品瓶中加入2 mL海水样品,分别加入0、20、50、100、200和500 μL HNO3,旋紧瓶盖放置2 h后,加水至25 mL,再分别加入150 μL 20%SnCl2溶液。配制0、20、50、100、200、500 pg的Hg做标准曲线,采用原子荧光检测器进行测定。从图3可以看出,当HNO3加入量为100 μL时,样品含量测定结果最高。当加入量大于100 μL时,汞含量测定结果稍低并趋于稳定,因此后续选择HNO3的加入量为100 μL。
2.5 消解时间优化
消解时间直接影响测定结果的准确度,因海水样品中可能含有少量甲基汞,因此以国际比对海水样品考察了不同消解时间对测定结果的影响。向2 mL海水中加入100 μL HNO3,分别放置0、10、30、60、120、300、1 020 min,完成后分别加超纯水至约25 mL,再分别加入150 μL 20%SnCl2溶液后,采用原子荧光检测器进行测定,同时做空白实验。从图4可以看出,当消解时间大于2 h后,Hg含量测定结果趋于稳定,后续实验选择消解时间为5 h。
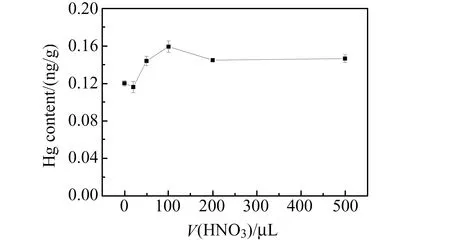
图3 HNO3加入量对测定结果的影响Fig.3 Effect of HNO3 volume on Hg detection

图4 消解时间对测定结果的影响Fig.4 Effect of digestion time on Hg detection
2.6 消解效果验证
首先测定HNO3消解5 h后样品的浓度为0.1560±0.0124 ng/g(平均值±标准偏差);然后参考EPA 1631规定的方法,向测定完成后的样品中加入100 μL BrCl,在60 ℃水浴中加热30 min,冷却后加入150 μL 30%盐酸羟胺,10 min后加入150 μL 20%SnCl2溶液进行测定,结果为0.127±0.004 ng/g。另外,将加入HNO3后的样品在60 ℃水浴中加热30 min,冷却后进行测定,结果为0.1567±0.0134 ng/g。上述三组实验每组平行3个样品,同时做空白扣除。从结果可以看出,加入HNO3放置5 h或者60 ℃水浴加热30 min,即可实现样品的完全消解。
2.7 同位素稀释质谱法测定结果
将总汞分析系统与ICP-MS联机,对经5 h消解后的样品采用单同位素稀释质谱法进行样品的测定,按照公式(1)计算测定结果。
(1)
其中,Ry、Rb、Rx分别为稀释剂、混合样品和海水样品中的同位素比值,my和mx分别为稀释剂和样品的质量,cy为稀释剂浓度,Mi为同位素i的摩尔质量。
为准确测定流程空白,同样采用同位素稀释质谱法进行空白的测定,而不是采用直接扣除信号的方式。样品和空白各测定6次,以空白测定值标准偏差的3倍作为检出限,经计算检出限为3.7 pg/g;以空白测定值标准偏差的10倍作为定量限,则吹扫捕集-同位素稀释电感耦合等离子体质谱法的定量限为13 pg/g。国际比对海水样品中总汞测定值为0.1607 ng/g,相对标准偏差为4.11%。为进一步验证方法,分别向渤海海水和舟山岛附近海水中加入汞标准溶液,使加入的浓度分别为31.0 pg/g和49.7 pg/g,进行加标回收率测定。3个海水样品测定结果和加标回收率见表1,国际比对海水样品测定谱图见图5。从表1可以看出,国际比对海水样品总汞含量较高,渤海海水测定结果略高于检出限,舟山岛附近海水中总汞含量低于检出限。从样品保存条件也可以看出,采集后进行酸化处理有利于汞的保存,但仍不能排除塑料桶放置对于汞的吸附。国际比对海水样品在保持低pH值的同时,采用稀的金溶液处理塑料包装瓶,可能对于汞的稳定起到了一定的作用。在30 pg/g和50 pg/g加标水平上,海水样品的加标回收率分别为98.1%和104%。
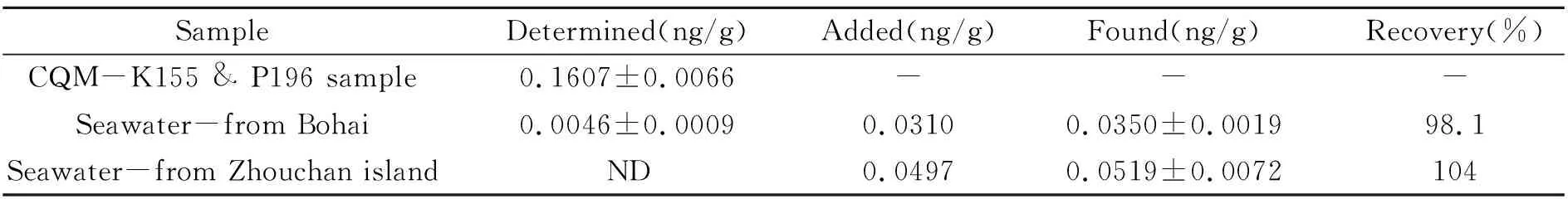
表1 测定结果及加标回收率(n=6)
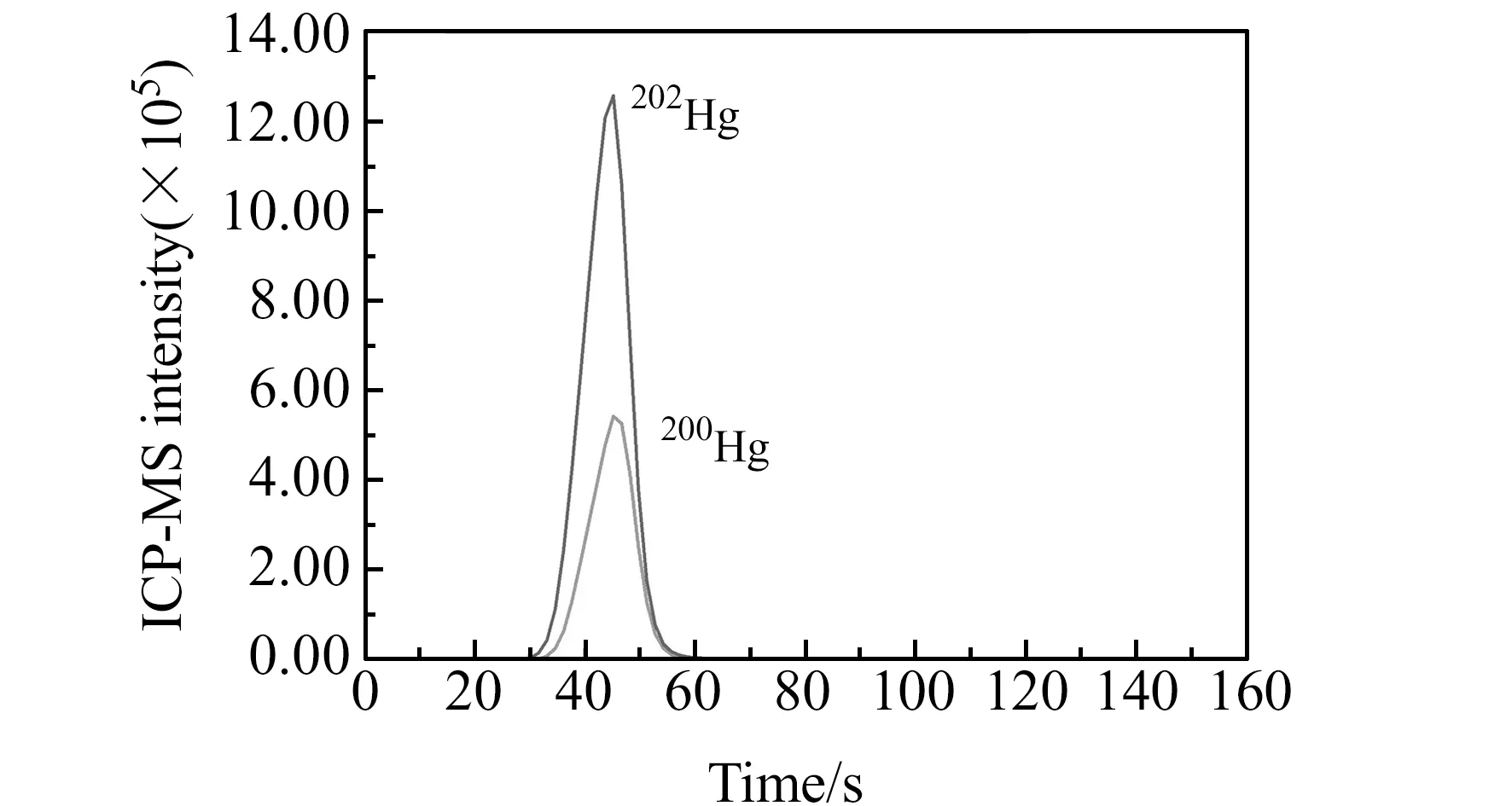
图5 海水比对海水样品ICP-MS信号图Fig.5 ICP-MS signal of CCQM comparison seawater sample
2.8 不确定度评定
为进一步评估该方法的计量学特性,针对国际比对海水样品,分别对稀释剂、流程空白、混合样品中同位素比值测定、样品称量等因素引入的不确定度分量进行评定,测量结果的相对扩展不确定度为4.69%(k=2),其主要来源为样品测定结果的标准偏差、混合样品中同位素比值测定、稀释剂本身和空白测定,各不确定度分量评定结果见表2。
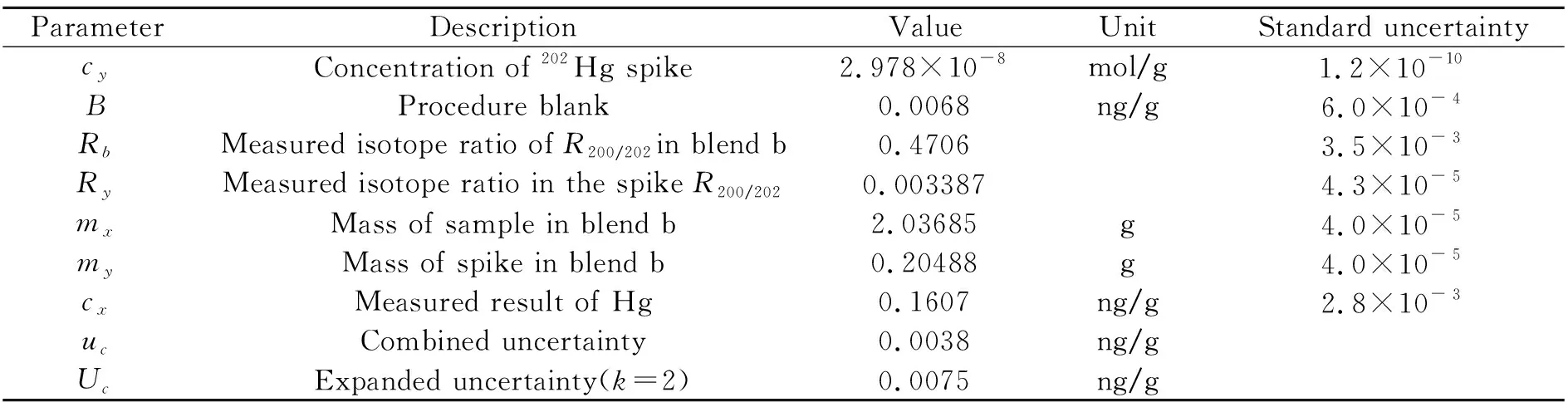
表2 测定结果的不确定度评定
3 结论
将吹扫捕集-原子荧光与电感耦合等离子体质谱联用,通过吹扫捕集-热脱附实现了汞与海水高盐基体的分离,金柱捕集进一步提高了灵敏度;以原子荧光检测器实时观测系统本底及清洗效果,同位素稀释质谱法作为潜在的基准测定方法,保证了测定结果的高准确度和良好的精密度,同时也避免了外标法在前处理过程中由于盐效应导致的样品与标准曲线中总汞吸附、脱附效率不同对测定结果的影响。HNO3进行样品消解、以加热或放置的方式即可实现总汞的消解,方法简单,避免了多种试剂加入对样品的汞污染。该方法准确度好、精密度高,适用于常规海水和国际比对、海水标准物质等样品中痕量汞的高准确度测定和定值。
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年21期)2021-08-30
食品安全导刊(2021年21期)2021-08-30
科学与财富(2021年33期)2021-05-10
分析化学(2018年4期)2018-11-02
食品界(2018年8期)2018-09-03
中国测试(2018年4期)2018-05-14
分析化学(2017年12期)2017-12-25
分析化学(2017年5期)2017-06-21
同位素(2014年3期)2014-06-13
