
掺杂CeO2光学性质和电子结构的第一性原理研究
2021-05-14 02:49:02申家欣郑浩然
华北科技学院学报 2021年1期
李 旭,申家欣,郑浩然
(华北科技学院 机电工程学院,北京 东燕郊 065201)
0 引言
在半导体工业中,基于杂质元素掺杂来实现宽禁带半导体吸收光谱的拓展,是提高一些本征材料半导体性能最有效的途径之一。目前,已经有许多关于各种材料的研究报道[1-2]。例如锰掺杂二硅化铬电子结构和光学性质的研究[3],不同元素掺杂ZnO电子结构和光学性质的计算研究报道[4],以及不同元素掺杂MgF2和Mg2Si电子结构和光学性质的第一性原理研究报道[5-8]。二氧化铈材料在玻璃行业中用作紫外线吸收剂,也可以作汽车尾气净化剂的助催化剂。近年来的研究发现CeO2材料可以作为固体氧化物燃料电池的氧离子导体的主要基体材料。二氧化铈(CeO2)是一种典型的稀土氧化物半导体材料,它的带隙比较宽,因环境或者温度条件的改变而容易发生相变,在各种催化剂或者添加剂等方面应用广泛。因为二氧化铈的禁带宽度有点大,所以在可见光范围光催化能力相对较弱。通过杂质原子对二氧化铈的掺杂,可以改善材料性质,继而扩展其应用领域。但是在CeO2方面的计算和分析还少见报道。
根据原子核和电子相互作用的原理及其基本运动规律,运用量子力学原理,从具体要求出发,经过一些近似处理后直接求解薛定谔方程的算法,习惯上称为第一性原理。本文采用基于第一性原理的计算法方法研究了Fe和N共掺杂CeO2的晶体结构,理论计算了光学性质和电子结构,比较分析Fe、N单掺杂以及共掺杂对CeO2光催化性能的影响,发现N单掺杂能提高太阳光中可见光吸收能力,而且在与金属共掺杂情况下能力提升效果更为显著。
1 计算模型及方法
采用Materials Studio软件中的CASTEP模块进行计算,利用基于第一性原理的平面波密度泛函(DFT),在广义梯度近似法(GGA)下,采取PBE泛函来确立交换—关联势。为了便于结果的分析比较,各结构体系在计算时采用相同的参数进行计算。电子-离子间的交互作用可以用采用超软赝势(ultrasoft pseudopotentials)更加合适,平面波截止能(cutoff energy)设置为340 eV, 设定自洽场(SCF)能量收敛精度为2.0×10-6eV/atom,能量收敛标准设为5×10-5eV/atom,原子间相互作用力收敛精度设置为0.5 eV·nm-1,晶体内应力收敛标准0.1 GPa,最大位移为2.0×10-4nm,布里渊区积分k的网格大小设置为1×2×2。首先构建2×1×1的超晶胞;然后再构建Fe和N在单掺杂情况下的体系结构及Fe原子和N原子同时掺杂的结构模型,掺杂原子的位置及晶胞的大小如图1所示。对以上四种结构模型进行结构弛豫,计算其各自的能带结构,态密度和光学性质,绘出能带、态密度、光吸收谱图,并对结果进行分析讨论和比较。
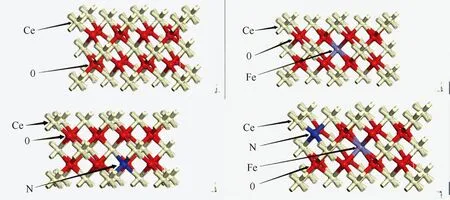
图1 未掺杂CeO2和各种掺杂CeO2结构模型
二氧化铈(CeO2)晶体结构同氟化钙(CaF2)类似,都是萤石立方结构。其原胞晶格常数为a=b=c=5.411Å,α=β=γ=90°,属于Fm3m空间群,原子分布是面心立方。如图2所示,铈和氧元素的配位数分别为8和4,每个氧离子(O2-)周围配位有4个铈离子(Ce4+),每个铈离子(Ce4+)周围配位有8个氧离子(O2-)。从晶胞结构看,二氧化铈含有八面体间隙,对于离子的扩散和掺杂具有良好的条件,因此很容易形成点、面、空位等缺陷。这些缺陷的增加可以提高体系的混乱度,使熵值增加,从而降低吉布斯自由能,体系的稳定性得到提高。
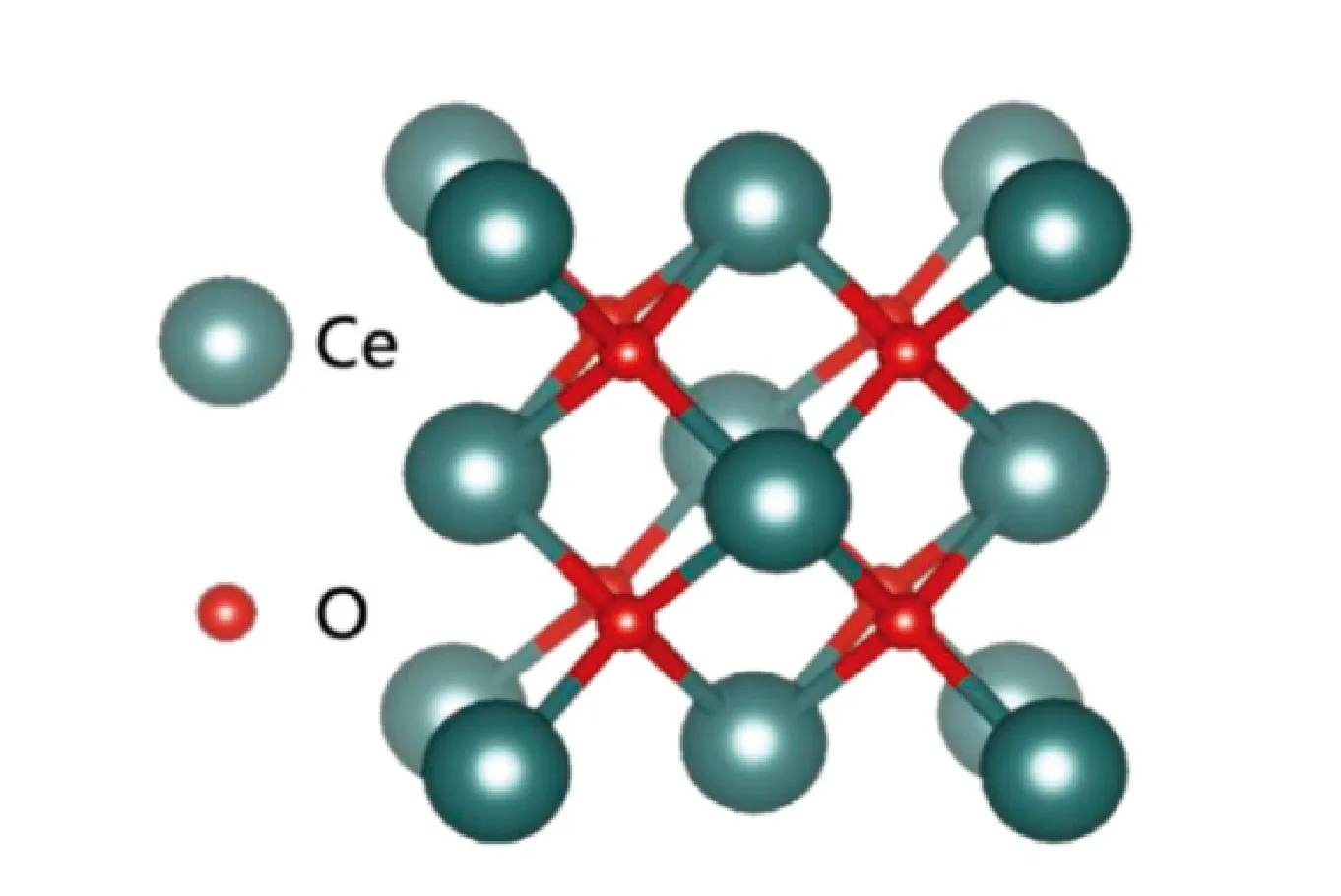
图2 CeO2结构类型
2 计算结果与讨论
2.1 晶体结构
首先,对2×1×1结构未掺杂CeO2、Fe单掺杂、N单掺杂和Fe/N共掺杂四种模型进行结构优化,优化后其晶胞参数如表3.1所示。从表中可知,掺杂后的晶胞体积分别为168.49Å3,168.50Å3,173.42Å3,对比掺杂前后的体积可以得出,掺杂导致了体积发生膨胀。由于N的离子半径比O略大,因此N替位O后形成的Ce-N键比Ce-O长,致使掺杂体系发生晶格畸变,体积变大。Fe掺杂情况下,在优化完后的结构体系中明显可以看到Fe-O键键长发生了变化,相比于原来的Ce-O键长,有的变长,有的变短,晶格发生了畸变。
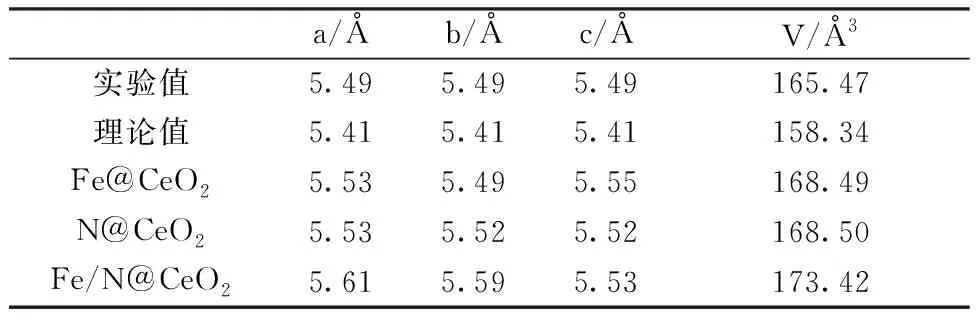
表1 掺杂前后CeO2的晶格常数和体积
2.2 态密度及能带结构的分析
如图3所示,图3(a)是未掺杂的CeO2的态密度图,能量等于零的位置是费米能级。由图中分态密度曲线分布情况可以看出,在费米能级附近的价带中,Ce原子的5d和4f轨道以及O原子的2p轨道虽然峰值不同,但都有存在,表明了Ce原子的4f轨道部分被占据了。从曲线的宽度和峰值高低分布差异,可以看出未掺杂的CeO2价带顶大部分都是由O原子的2p轨道贡献,也有少量的Ce原子的5d和4f轨道存在;导带底是由Ce原子的4f轨道贡献。图3(b)是N原子掺杂后的态密度图,在N原子掺杂后,从总态密度和分态密度的曲线形状和费米能级位置的区别可以看出,能带整体在向左边低能量方向移动,能带移动后电子价态更稳定,得电子更容易,CeO2价带的氧化能力增强,有利于光催化能力的提升。价带主要由N的2p和O的2p轨道,以及少量的Ce的5d轨道组成。掺杂后同样出现了杂质能级,由Ce原子的4f轨道和N的2p轨道共同作用形成。在图3(c)中,掺杂了Fe原子,从态密度图中可以看出,Fe原子的3d轨道在费米能级上下附近贡献,所以价带主要由Fe的3d轨道和O的2p轨道,以及少量的Ce的5d轨道组成,Fe原子的3d轨道与其他价带顶附近的轨道杂化,产生杂质带。导带主要Ce原子的4f轨道和Fe的3d轨道组成,轨道杂化产生杂质能级,掺杂后出现了杂质能级在费米能级附近。图(d)是Fe-N原子共掺杂CeO2的态密度图,价带主要由Fe的3d轨道、O的2p轨道、N的2p轨道以及少量的Ce的5d轨道组成,所以价带顶在费米能级附近的杂质带更加复杂,由杂质原子的轨道和O的2p轨道、少量Ce的5d轨道共同作用而产生。
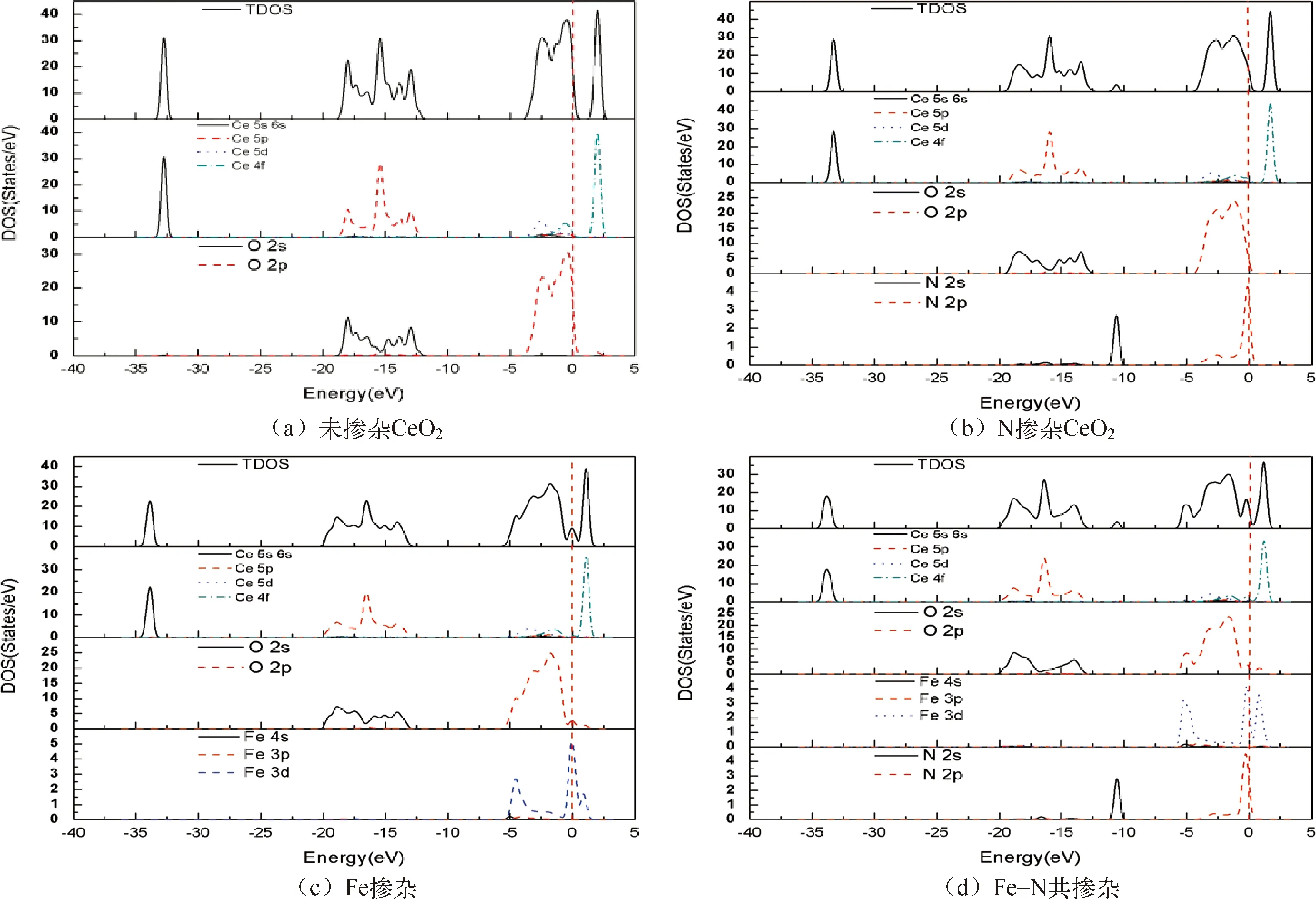
图3 未掺杂和掺杂后的CeO2态密度图
能带结构如图4所示,图4(a)是未掺杂CeO2的能带结构图,带隙是1.703 eV,导带顶为2.056 eV,导带底为1.703 eV。图4(b)是Fe掺杂CeO2的能带结构图,根据Fe掺杂的态密度图中各个原子的轨道贡献,在费米能级附近上下方出现三条比较孤立能带,这三条能带应该是杂质能级产生的。图4(b)的导带底为0.816 eV,导带顶为1.225 eV,除去三条杂质能级,价带顶为-0.72 eV。所以带隙为1.536 eV,相比未掺杂的CeO2带隙,掺杂过后带隙变窄了。掺杂后导带底下降了0.574 eV,导带的宽度增加了0.056 eV。图4(c)是N掺杂的能带图,也产生了杂质能级,在费米能级附近应该有三条杂质带,其中一条与费米能级相交,它的导带底为1.536 eV,导带顶为1.832 eV,除去三条杂质能级,价带顶为-0.268 eV。所以带隙为1.804 eV,相比未掺杂的CeO2带隙,掺杂过后带隙变宽了。掺杂后导带底下降了0.167 eV,导带的宽度也减少了0.057 eV。图4(d)是Fe和N的共掺杂能带结构图,从图中可以看出,费米能级附近应该有七条杂质带,费米能级上面有两条,下面有五条,所以它的导带底为0.959 eV,导带顶为1.34 eV,价带顶为-0.81 eV。所以带隙为1.769 eV,相比未掺杂的CeO2带隙,掺杂过后带隙变宽了。掺杂后导带底下降了0.744 eV,导带的宽度增加了0.028 eV。
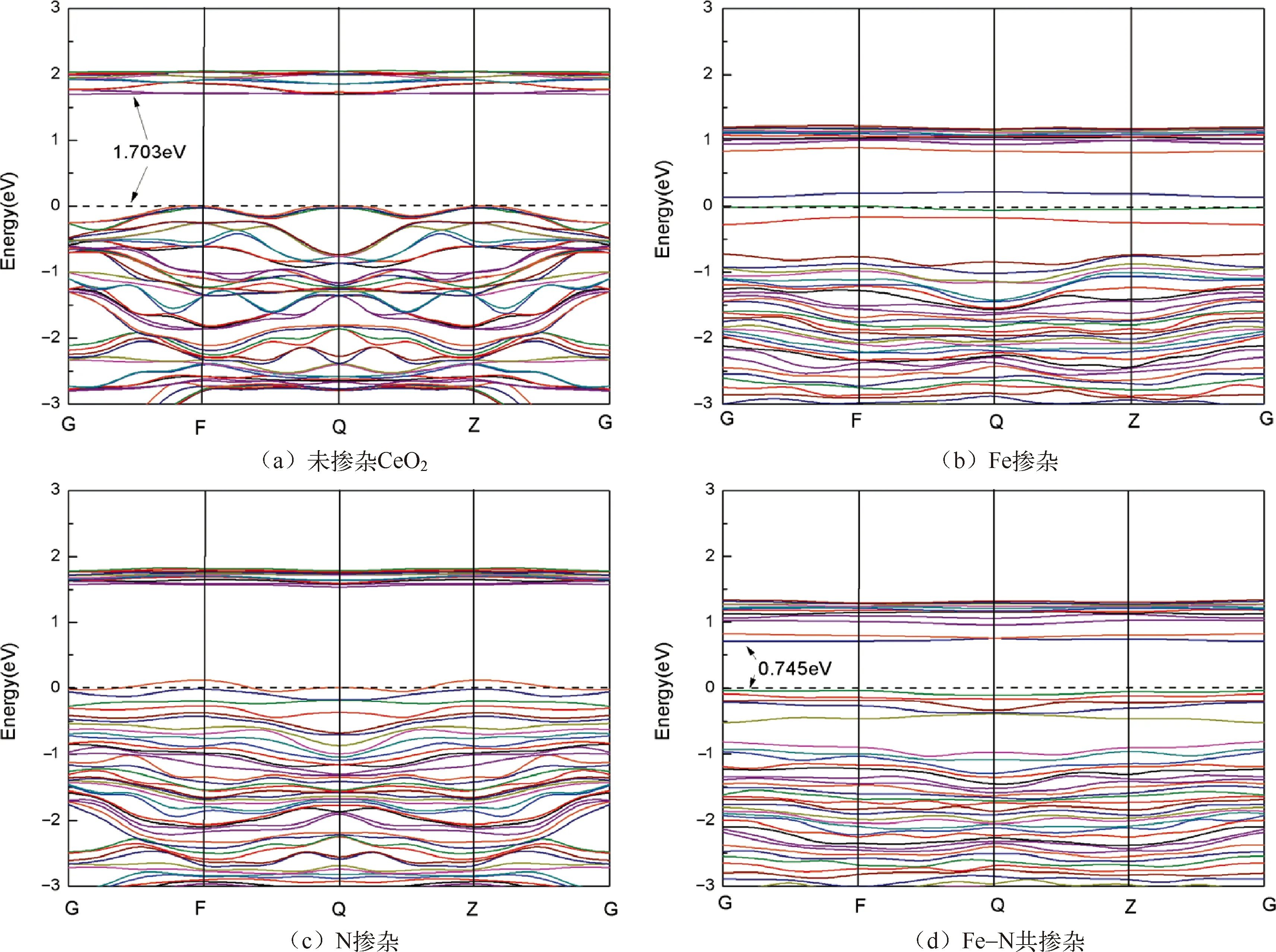
图4 未掺杂和掺杂后的CeO2能带结构图
综上所述,共掺杂带隙变宽,导带下移,体系变得更加稳定,金属掺杂会导致导带变宽,带隙变窄,非金属掺杂作用效果相反,但金属掺杂在费米能级上下都会产生杂质能级,使得价带电子更容易激发到导带,有利于提高其可见光的光催化性能。
2.3 光学性质
如图5所示,四种结构在同一X坐标轴上的吸收曲线情况,未掺杂CeO2的吸收曲线在可见光区域有一峰值,其他三种掺杂情况的曲线峰值都发生了蓝移,掺杂情况的吸收曲线都比未掺杂CeO2情况的曲线高,在三种掺杂情况之中,Fe和N两种掺杂曲线高度相当,在150~250 nm,350~680 nm波长区间,Fe掺杂的吸收曲线高于N掺杂的曲线,除此之外其他区间,N掺杂的曲线高于Fe掺杂的。Fe-N共掺杂的曲线除了在大约450~550 nm区间略微低于Fe单掺杂的曲线,其他区间都是高于其它三条曲线的。共掺杂吸收曲线有两个峰值,大约在200 nm和350 nm附近。可见,在掺杂情况下,产生的杂质能级起到了一个台阶的作用,在光催化情况下,电子从价带跃迁到杂质能级,再从杂质能级跃迁到导带。掺杂原子轨道的杂化,导致杂化能级的出现,拓展了可见光吸收范围。这三种掺杂的情况都提高了CeO2的光催化性能。
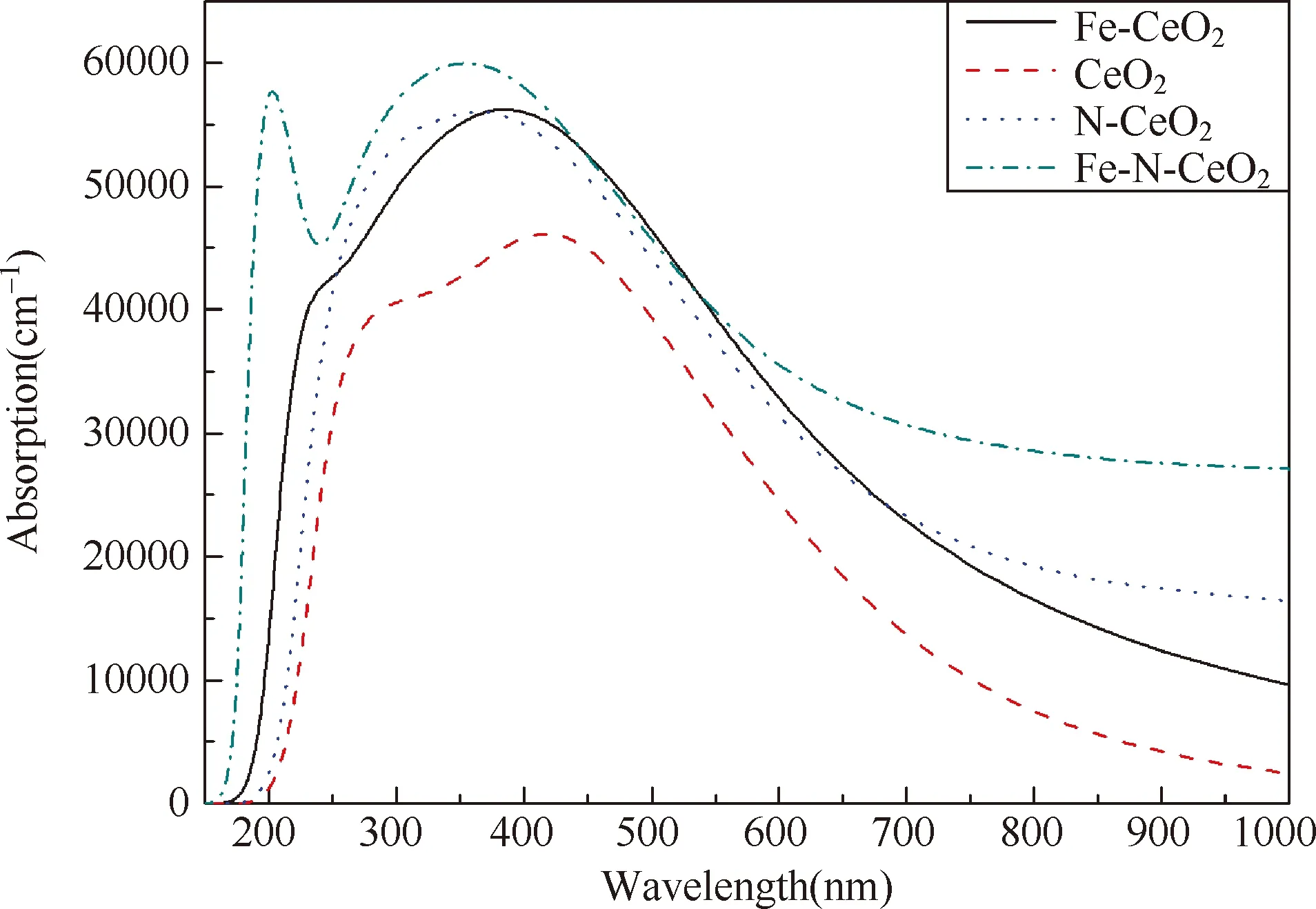
图5 掺杂和各种掺杂后CeO2的光吸收曲线
3 结论
(1) Fe和N元素单掺杂以及共掺杂后,晶胞都发生了畸变,体积变大,Fe掺杂后畸变程度最小,共掺杂畸变程度最大。
(2) 三种情况的掺杂后,导带均下移,导带变宽,密度减小;Fe和N单掺杂后带隙窄,共掺杂后带隙变宽,均产生了杂质能级,易形成P型半导体。
(3) 三种掺杂情况的光吸收曲线都比未掺杂的曲线高,都可以提高CeO2的光学性能;共掺杂的光吸收曲线有两个峰值,它的曲线最高;N单掺杂的曲线比Fe单掺杂在红外区高,如此可见共掺杂对CeO2的光学性能改善相对比较明显。
猜你喜欢
吉首大学学报(自然科学版)(2023年6期)2023-12-22 08:18:20
装备维修技术(2021年36期)2021-10-25 13:21:04
弹箭与制导学报(2021年3期)2021-07-30 02:56:52
数学物理学报(2019年6期)2020-01-13 06:08:24
发明与创新·小学生(2019年12期)2019-12-05 06:02:28
发明与创新(2019年47期)2019-11-21 01:16:18
网印工业(2019年4期)2019-05-21 06:41:58
重型机械(2019年2期)2019-04-28 11:52:04
吉首大学学报(自然科学版)(2018年3期)2018-07-03 03:14:12
Chinese Journal of Chemical Engineering(2017年5期)2017-05-28 10:22:54
