
核磁共振波谱法结合氧弹燃烧测定茶叶中的总氟
2021-05-10 06:47朱正伟朱松松王会霞
食品与机械 2021年4期
周 密 韩 智 朱正伟 朱松松 王会霞 江 丰 朱 芊
(1. 湖北省食品质量安全监督检验研究院,湖北 武汉 430075;2. 湖北省食品质量安全检测工程技术研究中心,湖北 武汉 430075)
氟是人体重要的微量元素之一,参与人体的许多生命活动[1]。氟适量摄入有益于人体骨骼和牙齿的钙化,增加骨骼的强度并维持口腔健康[2-3],但摄入过量则会导致全身慢性蓄积性中毒,形成“地氟病”,轻者形成氟斑牙,重者造成氟骨病,严重影响生长发育,危害身体健康[4-5]。茶树[Camelliasinensis(L.) O. Kuntze]有较强富集氟的能力[6-8],叶片是茶树富集氟的主要器官,尽管不同质量、不同产地、不同品牌的茶含氟量不同[9],但随着茶叶叶龄的增加,叶片中氟含量也逐渐升高[10-14],老叶含氟量更是高出嫩叶近10倍[15]。随着近年来消费提档升级,有学者[16]将超微茶粉直接开发成固体饮料,提高茶叶中有效成分的利用率,以满足市场多元化需求,但缺少对该类产品中总氟含量的关注,如若生产商使用老叶或劣质茶叶为原料加工生产,必定增加食用人群对于氟的摄入量,对身体健康构成威胁。
茶叶中氟的测定前处理方法主要有浸泡法[17]、灰化碱熔法[18]和氧弹燃烧法[19-20]。现行国标[17]中茶叶氟的测定方法主要是针对水溶性氟,但植物细胞内的氟难以被完全提取,无法准确反映产品中总氟含量;灰化碱熔法在高温、开放的体系中进行,处理时间长达3~5 h,氟易损失[20];氧弹燃烧使茶叶(粉)样品在密闭富氧的条件下反应,将氟元素全部转化为无机态氟[20],样品损失小,适合茶叶总氟含量测定[19]。
定量核磁共振技术(quantitative nuclear magnetic resonance,qNMR)已被广泛用于天然产物表征[21-22]、参考物质质量控制[23-24]和药物分析[25-26]中,其中,核磁共振氟谱(19F quantitative nuclear magnetic resonance,19F qNMR)具备化学位移范围大、灵敏度高、结构近似的化合物不易出现峰重叠[27]等优势,在定量分析中应用广泛。研究拟选择19F qNMR技术,以三氟乙酸为内标,测定茶叶中氟含量,可为茶叶及其制品中总氟的数据监控提供技术支撑。
1 材料与方法
1.1 材料与仪器
1.1.1 主要仪器设备
全数字化超导核磁共振谱仪:AVANCE Ⅲ 600 MHz型,德国Bruker公司;
超纯水系统:Purelab Chorus型,英国ELGA公司;
刀式捣磨仪:GM 300型,德国Retsch GmbH公司;
强制对流通用烘箱:UF 110 PLUS型,德国Memmert公司;
电子天平:ME 204型,梅特勒—托利多仪器(上海)有限公司;
粉末压片机:FW-5型,天津市拓普仪器有限公司;
卤素分析前装置:WBLS-1型,含充氧装置、点火装置,万邦仪表科技有限公司;
恒温振荡水浴锅:WNB 29型,德国Memmert公司;
超声波双频清洗机:SB25-12DTS型,宁波新芝生物科技有限公司。
1.1.2 材料与试剂
重水:纯度99.9%,Cambridge Isotope Laboratories, Inc;
高纯氧:纯度99.999%,四川天一科技股份有限公司武汉供气分公司;
氟化钠、碳酸铵、柠檬酸钠、乙二胺四乙酸二钠(EDTA-2Na)、三氟乙酸:分析纯,上海阿拉丁生化科技股份有限公司;
测试茶叶样品:湖北省食品质量安全监督检验研究院;
茶叶(绿茶粉)中氟质控样品(P24307):广州谱恩科学仪器有限公司。
1.2 试验方法
1.2.1 谱仪测定条件 在前人[28]的基础上,根据仪器硬件条件,对谱仪测定条件适当修改。脉冲序列为zgfhigqn,采样点数64 000,采样时间0.961 2 s,扫描次数32次,谱宽34 091 Hz,观察道中心频率偏置-56 468 Hz,去耦道中心频率偏置4 801 Hz,脉冲宽度12.0 μs,测定温度298 K。采用t1irpg脉冲序列,确定谱仪的测定参数延迟时间d1。
1.2.2 内标物选择及氟标准曲线 称取0.200 0 g三氟乙酸于100 mL容量瓶中配制成含氟当量为1.0 mg/mL的三氟乙酸溶液,将氟化钠于120 ℃烘干2 h,准确称取0.221 0 g,加水溶解,定容至100 mL,摇匀,浓度为1.0 mg/mL,进一步配制质量浓度为2.0,4.0,8.0,12.0,20.0 μg/mL溶液,均含氟当量为10.0 μg/mL 的三氟乙酸溶液。精密移取540 μL标准溶液,60 μL重水于核磁管中,混匀。按1.2.1所述方法采集19F NMR谱图,考察标物三氟乙酸与目标物氟标准溶液的分离情况,确定内标物;以目标物、内标物峰面积之比为纵坐标,目标物、内标物质量浓度之比为横坐标,考察线性关系。
1.2.3 样品前处理 参照文献[19—20],试样经捣磨仪粉碎、过40目筛,于80 ℃烘干至恒重,称取约0.50 g茶叶试样,压片后放入氧弹坩埚中,于氧弹反应釜内加入15 mL 20 mmol/L碳酸铵溶液作为吸收液,拧紧氧弹盖,缓慢充入3.0 MPa纯氧,将氧弹置于振荡水浴中,点火燃烧灰化,充分反应后开启放气阀,加入金属螯合剂,用温水少量多次润洗、超声,定容至50 mL,过0.45 μm滤膜,待测。按1.2.1所述方法采集核磁氟谱,比较加入不同金属螯合剂(柠檬酸钠溶液、EDTA-2Na溶液、柠檬酸钠—EDTA-2Na混合溶液,均含氟当量为10.0 μg/mL的三氟乙酸)对氟谱信号的影响。
1.2.4 数据处理及计算公式 使用Bruker Topspin 3.5软件对谱图进行相位和基线校正,对内标及目标峰进行峰面积积分,按式(1)计算积分结果。
(1)
式中:
X——待测物含量,mg/kg;
m——称样量,g;
mstd——每50.0 mL含有内标物的质量,μg;
Mx——待测物分子量;
Mstd——内标分子量;
Ax——待测物定量峰信号面积;
Astd——内标物定量峰信号面积;
nx——待测样品包含的氟原子数,按F-计为1;
nstd——内标物包含的氟原子数,按CF3COOH计为3。
2 结果与分析
2.1 延迟时间d1的选择
使用NMR定量分析时,一定要保证选择足够长的延迟时间d1(≥5×T1),否则对结果影响很大[28]。试验通过t1irpg脉冲序列,测定三氟乙酸和氟化钠的延迟时间d1,分别为3.0 s和1.6 s,故使用三氟乙酸为内标测定样品中氟离子时,选择d1为15.0 s。
2.2 内标物及定量峰的选择
使用内标进行定量分析过程中,需要保证内标物质峰与待测物质峰良好分离。加入一定量的内标溶液及氟化钠标准溶液,加入10% D2O,于1.2.1所述条件进行19F NMR测试,测试谱图见图1。
图1中δ=-120的尖锐单峰为氟化钠信号峰,δ=-75的尖锐单峰为三氟乙酸信号峰,与文献[28]相符,被测物质的定量峰与内标物的定量峰完全分离,说明使用三氟乙酸作为内标具有可行性。
2.3 线性关系的确定
取质量浓度为2.0,4.0,8.0,12.0,20.0 μg/mL溶液(均含氟当量为10.0 μg/mL的三氟乙酸溶液)进行线性关系考察。以样品积分面积与内标物的积分面积比(S1/S2)为纵坐标,以样品的浓度与内标物的质量浓度比(C1/C2)为横坐标,结果见图2。
由图2可知,当样品质量浓度为2.0~20.0 μg/mL时,S1/S2与C1/C2呈良好的线性关系,回归方程为y=1.027 1x+0.037 8,回归系数R2=0.999 5,进一步说明使用三氟乙酸作为内标对样品中氟进行定量分析是可行的。
2.4 金属螯合剂的选择
氟离子作为性质活泼的阴离子之一,易与Ca2+、Fe3+、Mg2+、Al3+等金属阳离子形成络合物从而影响测定,GB 19965—2005通过添加柠檬酸钠屏蔽金属离子,也有学者[29]通过添加EDTA-2Na络合金属Al元素。试验选取2.50%柠檬酸钠溶液和1.25% EDTA-2Na溶液作为金属螯合剂,考察金属螯合剂添加与否、不同金属螯合剂添加量对测定结果的影响。

图1 内标物与目标物的19F qNMR谱图
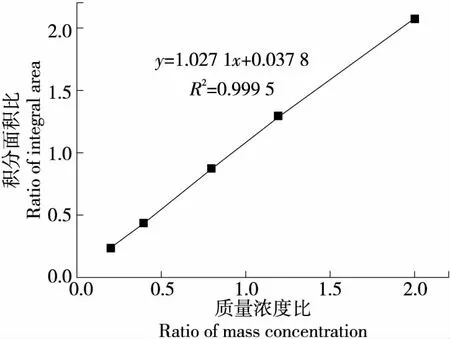
图2 积分面积与质量浓度线性关系
结果表明,在不添加金属螯合剂和只添加1.25% EDTA-2Na溶液5 mL时,经过氧弹燃烧后的样品在δ=-119处无目标峰出现,如图3(c)和图3(d)所示;图3(b)中,在添加2.50%柠檬酸钠溶液5 mL时,样品在δ=-119处出现目标峰,但峰型较差,定量困难;而在添加金属混合螯合剂(1.25% EDTA-2Na溶液、2.50%柠檬酸钠溶液)5 mL时,样品在δ=-119处目标峰峰型良好,如图3(a) 所示,通过增加扫描时间,即可对目标物质进行定量分析。这可能是由于不同螯合剂对不同价态金属螯合作用不同,进一步将性质活泼的络合态氟元素,从与金属络合的状态下释放出来。另外,增加金属螯合剂至10 mL 时结论一致,但不同金属混合螯合剂添加量对峰响应强度有一定影响。
进一步添加0.0,2.5,5.0,7.5,10.0 mL不同水平的金属混合螯合剂,比较目标峰响应响度,见图4。结果表明,未添加金属混合螯合剂时无目标峰响应,金属混合螯合剂添加至5.0 mL后响应强度趋于稳定,继续添加7.5,10.0 mL 金属混合螯合剂,信号响应强度无明显变化,说明在试样称样量为0.5 g水平下,金属混合螯合剂与溶液中的金属离子充分络合。故氧弹反应完全,添加5.0 mL金属混合螯合溶液为宜。
2.5 方法学考察
2.5.1 最低检测浓度 依据《中国药典》(2015版)第4部通则,以S/N≥3时确定最低检测浓度,在1.2.1所述条件下,当δ为-70~-130,测定时间为8 min 31 s时,最低检测浓度为2.0 μg/mL。
2.5.2 重复性试验 选取茶叶(绿茶粉)中氟质控样品(P24307)、砖茶样品,按1.2.3所述方法,重复测定6次,氟质控样品典型测试谱图如图5所示,定义三氟乙酸峰面积为1.000 0(原始积分数据3 568 081.9),目标峰氟离子面积为0.293 7(原始积分数据1 047 966.5)。测试结果如表1所示,其相对偏差为6.04%~6.98%,方法重复性良好。另外,茶叶(绿茶粉)中氟质控样品(P24307)中位值为319 mg/kg,|Z|≤2.0时测试值范围为258~380 mg/kg,测试值为301 mg/kg,测试结果满意。

图3 螯合剂对响应强度的影响
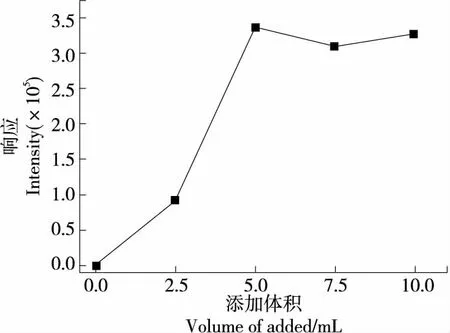
图4 不同体积螯合剂对响应响度的影响
2.5.3 精密度试验 随机选取茶叶样品,通过19F qNMR平行测定总氟含量6次,定义三氟乙酸峰面积为1.000 0,对目标峰积分,计算相对峰面积,6次积分面积结果相对偏差为2.50%,表明使用19F qNMR测定总氟含量具有较高的精密度。

图5 典型样品测试谱图
2.5.4 回收率试验 茶叶样品基质(1 226 mg/kg)中添加氟含量分别为200,400,2 000 mg/kg三水平的标准溶液,每个水平平行测定3次,计算回收率和相对偏差,结果如表2所示,加标回收率为96.3%~107.0%,相对偏差为4.20%~6.07%。
3 结论
采用氧弹燃烧对样品进行前处理,以三氟乙酸为内标,在无外部校准曲线的情况下,结合19F qNMR对茶叶中的总氟进行快速定量分析,填补中国使用19F qNMR测定茶叶样品总氟含量的技术空白,30 min内可完成单个样品分析,检测效率高,方法准确、稳定性良好,具有一定的实用性。在对茶叶样品总氟含量的分析测定中,可以考虑添加混合螯合剂,进一步释放与金属离子络合的氟元素。该方法在灵敏度上存在一定的局限,后续研究中可考虑优化谱仪脉冲条件、采用变温试验等方式来提高检测灵敏度。

表1 重复性试验

表2 回收率试验
猜你喜欢
环境科学研究(2022年10期)2022-10-19
中南民族大学学报(自然科学版)(2022年3期)2022-05-08
浙江化工(2022年1期)2022-02-19
中国土壤与肥料(2021年5期)2021-12-02
中国农业气象(2021年12期)2021-11-30
口腔护理用品工业(2021年4期)2021-11-02
小学科学(学生版)(2021年3期)2021-04-13
小哥白尼(趣味科学)(2020年9期)2021-01-18
中国科技纵横(2019年23期)2019-02-14
Coco薇(2015年5期)2016-03-29
