
两样本孟德尔随机化中工具变量对因果估计的性别特异性影响
——人体测量学性状与乳腺癌关系的实证研究*
2021-05-08 07:50徐州医科大学公共卫生学院流行病与卫生统计学系221004高一欣赵华硕
中国卫生统计 2021年2期
徐州医科大学公共卫生学院流行病与卫生统计学系(221004) 高一欣 曾 平 赵华硕
【提 要】 目的 研究两样本孟德尔随机化(mendelian randomization,MR)分析中性别特异工具变量对因果效应估计的影响。方法 利用全基因组关联数据以乳腺癌作为结局,以人体测量学性状(身体质量指数(BMI)、腰臀比(WHR)、腰围(WC)和臀围(HIP))作为暴露,采用两样本MR逆方差加权法估计因果效应;通过差异检验比较使用女性特异工具变量与性别合并工具变量的因果效应(OR值);进一步通过敏感性分析验证结果的稳健性。结果 使用性别合并工具变量的MR结果表明BMI/WC与乳腺癌风险存在因果关联(OR值分别为0.85(P=0.003)和0.87(P=0.020));剔除性别差异的工具变量后,每组OR值与未剔除前基本一致,但WC与乳腺癌的因果关联不再显著(P=0.069);使用女性特异工具变量的MR结果与使用性别合并工具变量结果相比,每组OR值均呈下降趋势;其中BMI/HIP与乳腺癌的因果关联效应大小发生了明显改变(P<0.05);例如BMI与乳腺癌因果关联的OR值由0.85下降至0.76。结论 工具变量的性别异质会对MR的因果效应估计产生实质影响,使用性别合并的工具变量可能导致有偏的因果关联。
众所周知,观察性研究中无论采用何种研究设计都难以避免众多已知或未知的混杂因素,从而会对因果推断的效应估计产生偏倚[1-2]。孟德尔随机化(mendelian randomization,MR)方法为解决这一问题提供了行之有效的途径;MR利用遗传变异作为工具变量估计暴露与结局之间的因果关联[3]。为确保MR的有效性,工具变量需满足三个核心假设[4]:(1)与暴露密切相关;(2)与任何影响暴露和结局的混杂因素不相关;(3)只能通过暴露影响结局,即不存在任何多效性效应。过去十几年来大规模全基因组关联研究(genome-wide association study,GWAS)已发现大量单核苷酸多态性(single nucleotide polymorphism,SNP)位点与数百种复杂疾病或性状(如血脂[5]、身体质量指数(body mass index,BMI)[6]、吸烟[7]和饮酒[8])存在关联[9],使得研究者能直接从GWAS中选择合适的SNP作为某种暴露的遗传工具变量[10-11]。
由于数据共享和隐私等限制,个体水平的GWAS数据不易获取,实际中往往利用相同群体的两个独立GWAS汇总数据探索暴露与结局的因果关联,即所谓两样本MR(two-sample mendelian randomization)[11-12]。另外,由于单个工具变量所能解释的遗传方差极其有限,为提高MR的检验效能往往使用多个独立的工具变量,然后采用类似meta分析的逆方差加权法(inverse-variance weighted,IVW)合并单个工具变量的因果效应估计值从而获得最终的效应估计[13-14]。总体而言,两样本MR简单且易于实施,为流行病学观察性研究因果推断提供了强有力的统计工具。
然而,实际中两样本MR分析依然面临诸多应用和方法上的挑战。首先,两样本MR要求暴露与结局的GWAS样本互不重叠,否则会产生有偏的因果效应估计[15]。其次,暴露与结局的GWAS人群应具有相似的种族特征,如均来自欧洲或亚洲人群[16]。除此之外,对某些特殊疾病(如女性乳腺癌,男性乳腺癌仅占约1%不在本文讨论之列),其暴露的工具变量会出现性别异质性(sex heterogeneity)问题。理论上,当采用两样本MR估计某些暴露因素(如BMI)与乳腺癌的因果关联时,从暴露GWAS中获取的工具变量效应大小理应是女性特有的。大量的GWAS曾报道很多人体测量学性状的遗传结构存在性别差异(表1);例如,研究者在多个与肥胖相关的基因座上发现了性别差异,其中大多数与腰臀比(waist-to-hip ratio,WHR)相关的SNP在女性中呈现出比男性更强的效应[17-19]。然而,很多研究者在面对类似问题时往往简单忽略工具变量性别异质性,这很可能会产生错误的因果推断结果[12,20]。另外,实际中GWAS协作组并没有公开发布性别相关的汇总数据而仅有男女混合的汇总数据,因此研究者不得不采用男女混合的工具变量。需要注意的是,这一问题是基于汇总数据的两样本MR分析,由于数据的可获得性而特有,在个体水平数据的因果推断分析中不存在。在这种情况下,应用者仍需对工具变量性别异质性可能产生的因果估计偏倚做出说明并进行敏感性分析。
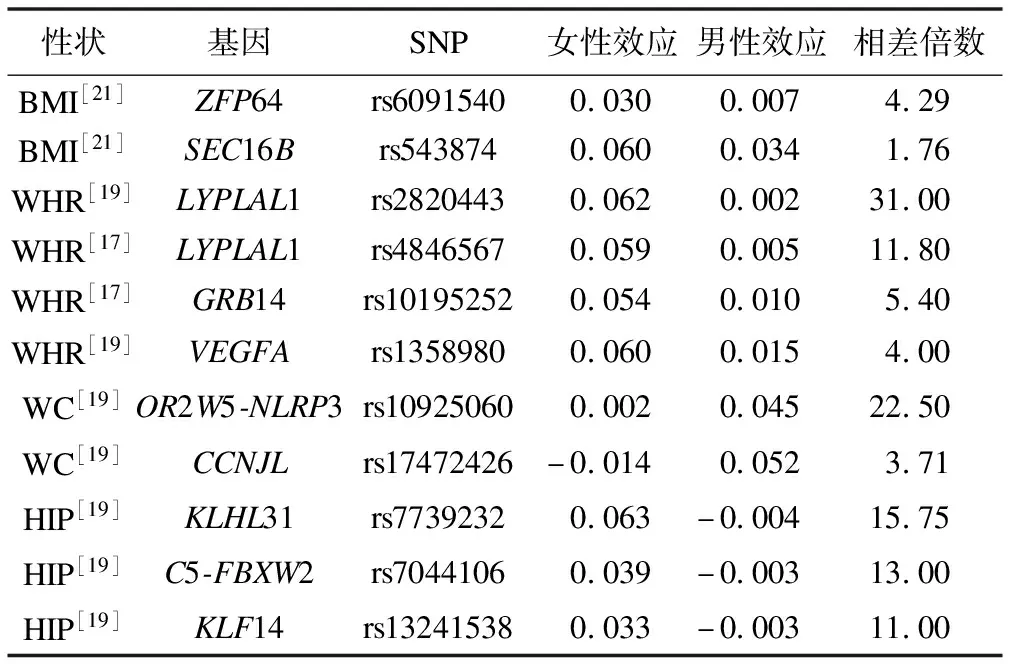
表1 GWASs报道的具有显著性别差异的基因和SNP
我们的文献综述显示大多数两样本MR分析尚未系统研究过工具变量性别异质性的问题。因此,本文以乳腺癌和四个人体测量学性状(即身体质量指数BMI、腰臀比WHR、腰围WC和臀围HIP)为例,说明工具变量性别异质性在MR分析中可能产生的一系列问题,并提出相应的分析策略和建议。
资料和方法
1.数据来源
鉴于人体测量学性状具有性别差异的客观证据[22-23],本研究从人体性状遗传研究联盟(GIANT)获得了四个人体测量学性状(即BMI、WHR、WC和HIP,后三种性状均对BMI加以校正)的性别合并和性别特异的GWAS汇总数据[19,21]。从国际乳腺癌研究联盟(BCAC)获得了女性乳腺癌的GWAS汇总数据[24]。所有个体均为欧洲血统(表2)。
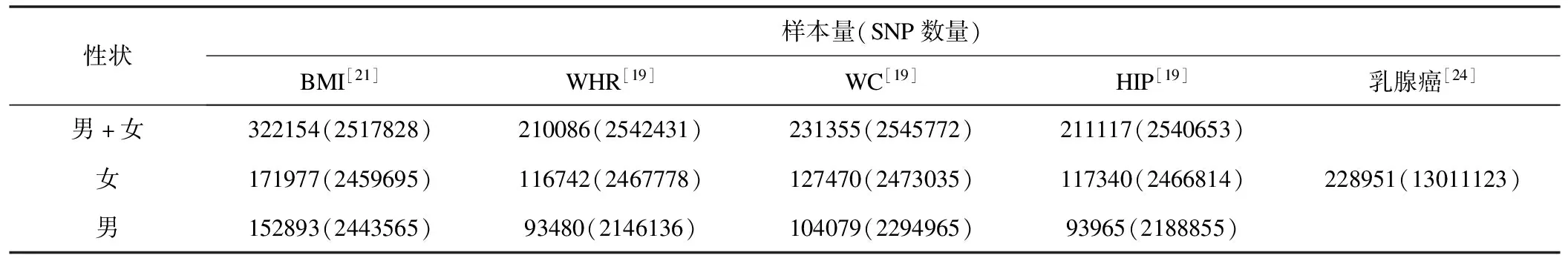
表2 MR分析中使用的GWASs数据信息
2.工具变量的选择
首先,从GWAS中直接筛选达到全基因组显著性水平(即P<5×10-8)的SNP作为每个人体测量学性状的工具变量(表3)。随后,根据男性和女性的效应估计以及标准误进行性别异质性检验,使用Cochran Q统计量以及Bonferroni校正P值(即0.05/给定人体测量学性状的工具变量数)评估每个工具变量的性别异质性。
为提供更全面的结果,根据以往研究[25],运用PLINK(版本v1.90b3.38)[26]的clumping程序生成另一组女性特异的工具变量(表3)。SNP的主要显著性水平和次要显著性水平均设置为5×10-8,连锁不平衡和物理距离分别设置为0.01和1Mb,以千人基因组项目中的503个欧洲个体基因型作为参考面板[27]。
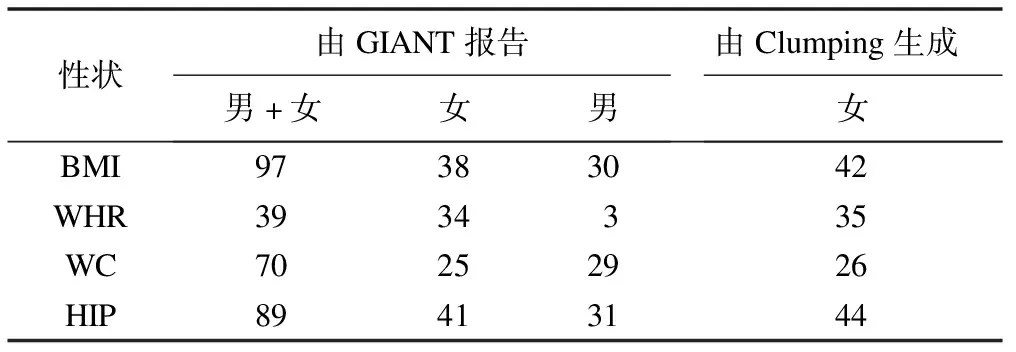
表3 达到全基因组显著性水平的工具变量数
3.因果效应估计
首先,利用性别合并的工具变量,采用两样本MR[28]中的IVW法[13,29]估计人体测量学性状与乳腺癌的因果关联。为检验性别异质性的工具变量对因果效应估计的潜在影响,通过剔除具有性别异质性的工具变量进行敏感性分析。其次,利用女性特异的工具变量再次实施两样本MR。采用差异检验比较使用女性特异工具变量与性别合并工具变量的因果效应
其中,d和SEd分别代表效应估计之差和标准误之差;ρ代表相关系数,我们的研究显示ρ基本在0.8以上,因此本文中设置ρ=0.8。
最后,若使用女性特异的工具变量确定了某种人体测量学性状与乳腺癌存在因果关联,则对其进一步实施加权中值法[30],极大似然估计[31],留一法(leave-one-out,LOO)[32],MR-PRESSO测试[33]和MR-Egger回归[34-35]等一系列敏感性分析验证MR结果的稳健性。
结 果
1.因果效应估计结果
(1)使用性别合并的工具变量
基于随机效应的IVW结果显示,BMI和WC每增加1个标准差,乳腺癌的OR值分别为0.85(95%CI:0.76~0.95,P=0.003)和0.87(95%CI:0.77~0.98,P=0.020)(表4)。同时,性别异质性检验发现了2个与BMI相关的SNP、17个与WHR相关的SNP、10个与WC相关的SNP以及4个与HIP相关的SNP。将这些SNP剔除后,与原始结果相比,OR与未剔除前基本一致(表4),但WC与乳腺癌风险的因果关联不再显著(P=0.069),表明具有性别异质性的工具变量的确会对因果效应估计产生影响。

表4 人体测量学性状与乳腺癌风险的因果关系(使用性别合并的工具变量)
(2)使用女性特异的工具变量
从结果可见,与原始结果相比,使用GIANT报告的女性特异工具变量后WC与乳腺癌风险的因果关联不再显著(P=0.057)(表5);同时,每种人体测量学性状与乳腺癌因果关联的效应大小均呈下降趋势,其中BMI/HIP与乳腺癌的因果关联效应大小发生了明显改变(P<0.05)(表5)。例如,BMI每增加1个标准差,使用性别合并工具变量的乳腺癌发病风险降低约15.5%(OR=0.85)(表4),而使用女性特异工具变量的乳腺癌发病风险降低约21.8%(OR=0.78)或23.7%(OR=0.76)(表5);表明使用性别合并的工具变量可能会导致有偏的因果关联。
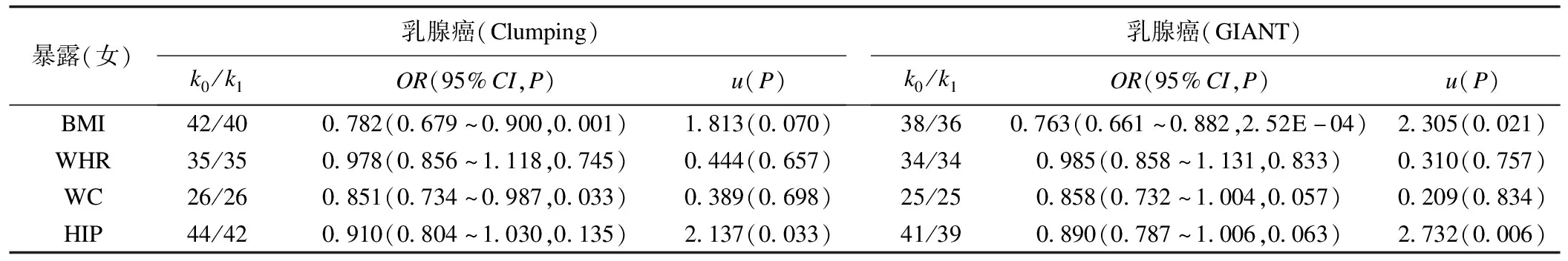
表5 人体测量学性状与乳腺癌风险的因果关系(使用女性特异的工具变量)
2.敏感性分析
由于表5显示BMI/WC与乳腺癌的因果关联显著,故对其进一步进行敏感性分析(图1)。加权中值法和极大似然估计与IVW法的结果近似。通过构建散点图(图2A1~C1),我们发现可能存在潜在的异常值工具变量。为检验这些异常值是否对因果效应估计产生影响,以由clumping生成的两个BMI异常值为例(即rs2229616和rs17024393),依次将其剔除后的效应(OR=0.79,95%CI:0.69~0.91,P=1.30E-03)和(OR=0.79,95%CI:0.69~0.91,P=1.20E-03)与同时将其剔除后的效应(OR=0.80,95%CI:0.70~0.93,P=2.48E-03)基本一致,且近似于未剔除前的效应大小(OR=0.78)。此外,LOO和MR-PRESSO表明,并不存在对因果效应估计产生实质性影响的工具变量。最后,由于MR-Egger回归的截距没有明显偏离于0,且漏斗图的因果效应点估计呈对称模式(图2A2~C2),进一步表明遗传多效性不会对因果效应估计产生偏倚。
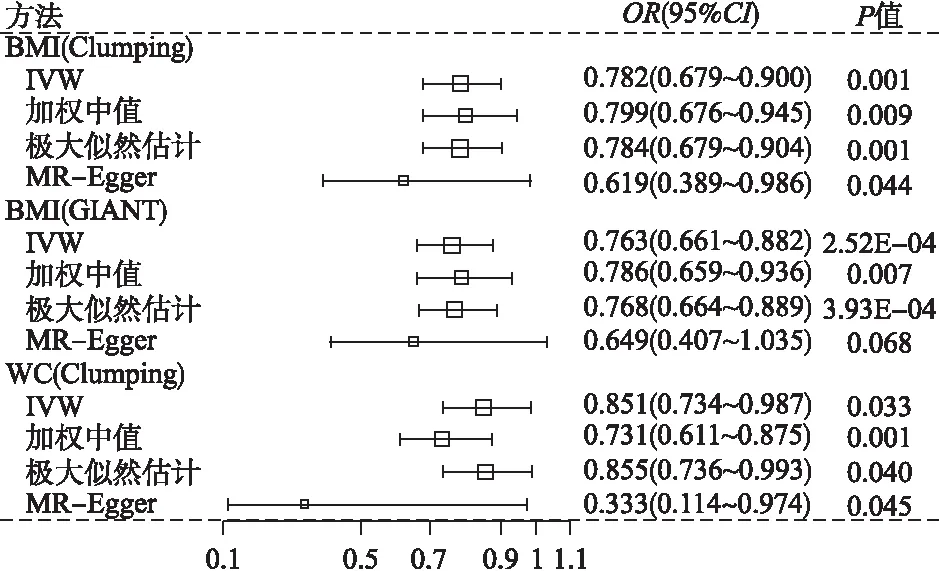
图1 敏感性分析
图1中MR-Egger为移除潜在异常值后的结果,Egger截距自上而下分别为0.009(95%CI:-0.006~0.025,P=0.239)、0.006(95%CI:-0.009~0.022,P=0.418)和0.033(95%CI:-0.002~0.068,P=0.066)。
图2散点图 A1~C1中的蓝线及漏斗图A2~C2中的垂直红线均表示IVW估计的因果效应,散点图中的红点代表潜在异常值。(A)使用40个由clumping生成的与BMI相关的女性特异SNPs,异常值由左向右依次是位于GNAT2上的rs17024393和MC4R上的rs2229616;(B)使用36个来自GIANT的与BMI相关的女性特异SNPs,异常值是位于GNAT2上的rs17024393;(C)使用26个由clumping生成的与WC相关的女性特异SNPs,异常值由左向右依次是位于VEGFA上的rs998584和CYCSP55上的rs1776897。
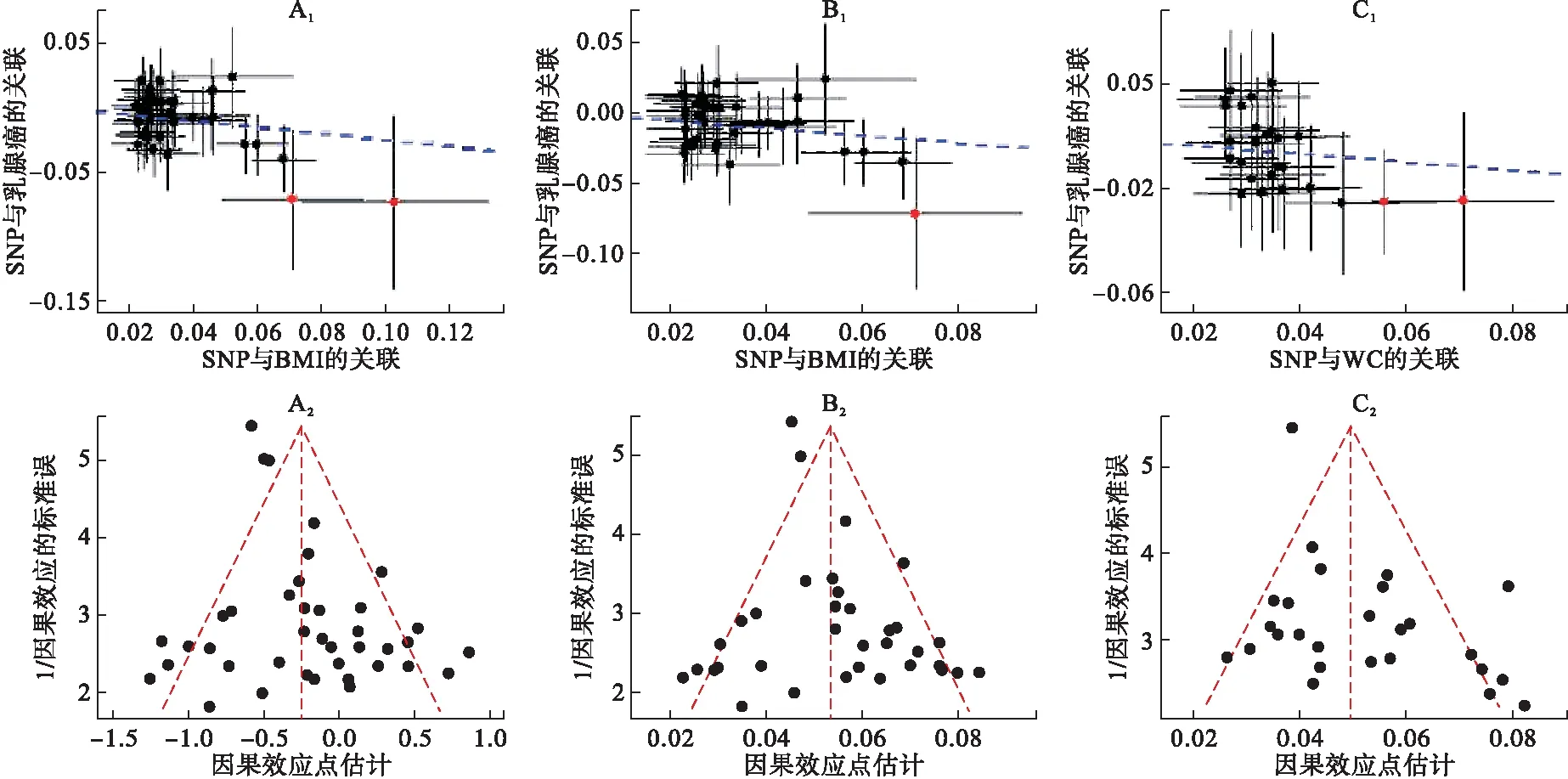
图2 散点图及漏斗图
讨 论
乳腺癌是全球绝大多数国家女性最常见的癌症,其发病率与死亡率均位于女性癌症患者首位[36]。迄今为止,乳腺癌的MR实例分析已多达61篇,MR的广泛应用为识别乳腺癌的复杂病因提供了极佳途径。然而,我们发现仅个别原始报告[37-40]中使用的工具变量效应大小是女性特有的(女性特异暴露如乳房大小、初潮年龄等的MR研究除外);尤其对于某些已被证实性别差异显著的性状如WHR等,在性别相关的汇总数据公开发布的情况下,绝大部分研究者依然简单地使用性别合并的SNP效应[41-42]且并未在文章中澄清这一点。两样本MR分析中使用性别合并指示变量事实上潜在假设性别之间不存在效应差异,而在实际中这一假设并不一定成立(例如表1的结果)。使用性别合并指示变量也潜在增加了暴露所在的GWAS样本量。在本文中,也即是假设男性样本可用于女性样本从而用于BMI等的关联检验,这将导致有更多的指示变量被选择和用于MR分析,在提高检验效能的同时也增加了指示变量间异质性的风险。
本研究借助人体测量学性状以及女性乳腺癌的大规模GWAS汇总数据,发现使用性别特异和性别合并的工具变量得到的MR结果存在实质的差别,且在因果效应估计方面存在偏差。在我们的实例研究中使用性别合并的工具变量会削弱暴露与结局的因果关联。据我们所知,本研究是首次利用实例数据探讨两样本MR分析中性别特异工具变量对因果效应估计的影响。
两样本MR的严格方法论认为,若两样本来自不同的人群,因果效应估计可能存在偏倚[1]。 MR仍可证明现有暴露与结局间是否存在因果关联,但因果关联的效应大小不能准确估计[43]。通过本研究发现,使用女性特异的工具变量,与使用性别合并的工具变量相比因果效应估计的大小呈下降趋势。当工具变量从男女混合的样本中提取,由于男性乳腺癌的稀缺性,加入了男性的效应即意味着减弱了原本存在的因果效应。本研究还注意到,单从数值看因果效应估计的下降幅度不大,我们认为这是合理的:若因果效应降幅明显,则意味着以人体测量学性状为典型的各类表型在男性和女性之间本就存在显著差异,而同一人群中不同性别的表型差异十分显著的现象并不多见[44-45]。因此,该结果证明,多数研究者未考虑工具变量的性别异质性,会导致MR的效应估计产生偏倚。
我们建议在类似的MR分析中应该首先查看原始论文以及出版物或联系原作者,明确是否可能在相同性别的人群中实施MR,同时分析时明确说明[39]。其次,当使用性别合并的工具变量时需进行性别异质性检验,移除显著性别差异的工具变量以实施敏感性分析;当使用性别特异的工具变量时需同步进行敏感性分析以确保结果的稳健性。最后,若无法获得指定的工具变量或无法完成上述敏感性分析,可借鉴Au Yeung[40]和Jiang[46]等人的文章,报告或讨论可能存在的偏倚,同时进一步考虑实施MR分析是否可靠。
总之,尽管使用公开汇总数据的MR在技术上是易于实现的,但仍应遵循MR的基本理论与原则。尤其在评估暴露因素与性别特异疾病的因果关联时,选择合适的性别特异工具变量可以在一定程度上减少因果推断的偏倚,使MR结果更加可信。
猜你喜欢
大学教育(2022年3期)2022-05-16
新世纪智能(数学备考)(2021年9期)2021-11-24
现代企业(2021年2期)2021-07-20
当代陕西(2019年15期)2019-09-02
学苑创造·A版(2018年11期)2018-02-01
读者(2017年5期)2017-02-15
邯郸职业技术学院学报(2016年2期)2016-02-27
华南农业大学学报(社会科学版)(2015年3期)2016-01-11
中国教育技术装备(2015年6期)2015-03-01
测绘学报(2015年8期)2015-01-14
