
基于苯并噻二唑的非共价构型锁定稠环小分子的合成与性能研究*
2021-05-07 12:03王英英秦红梅李宇翔孙道通杨建业
功能材料 2021年4期
王英英,秦红梅,王 梅,李宇翔,孙道通,杨建业
(西安科技大学 材料科学与工程学院,西安 710054)
0 引 言
近年来,有机半导体材料的研发和应用推动了有机光电器件的迅猛发展[1],特别是采用共价键偶连的新型稠环化合物取得了明显的进步,例如在有机光伏领域采用此类稠环化合物作为受体材料时器件的最大光电转换效率可以达到17%,这也是目前有机光伏器件的最高转换效率[2]。但此类稠环小分子化合物[3-5]存在一些缺点,如合成步骤复杂繁琐,不易纯化,且增加了制备过程和成本。因此,科研人员们开始将一部分目光转向了基于非共价键相互作用的稠环小分子材料[6-9]。通过将一些带有杂原子N、S、O、F等的基团引入到小分子中,利用分子内的S…O,N…H和O…H等非共价键[10-12]相互作用来增强整个分子骨架的平面性。采用这种非共价键相互作用的策略可以增强分子内电荷转移[13],进而拓宽光谱吸收,降低带隙,同时还有利于提高材料的电子迁移率[14-15]。
在众多有机共轭化合物中,苯并噻二唑(BT)基团具有一定的刚性平面结构、吸光系数高、溶解性好、良好的载流子迁移率、较强的吸电子能力、较高的氧化电位和较好的空气稳定性等优点,使得与其作为中心核所合成的小分子化合物具有一定的平面刚性和较好的载流子迁移率[2]。目前,基于苯并噻二唑的共价键型稠环小分子化合物,如:BT1[16]、BT2[17]、2CN-T-BT[18]、Y6[19]、BTP-eC9[2]等,已成功应用于有机半导体领域且展现出良好的应用前景。
基于以上,本文设计合成了基于苯并噻二唑(BT)为中心核,以并二噻吩为桥联,强吸电子单元(二氰基亚甲基)靛酮(IC)和5,6-二氟-3-(二氰基亚甲基)靛酮(IC4F)为端基的两个共轭非共价键非稠环小分子化合物(BT2T-IC、BT2T-IC4F)。随后,我们对所合成的两个小分子化合物的结构、电化学性能和光物理性能进行了表征和分析。
1 实验部分
1.1 实验与仪器
1H NMR和13C NMR:德国Bruker 400MHz核磁共振光谱仪;UV-Vis:日本SHIMADZU UV-3600 Plus测定;电化学工作站:上海晨华CHI600E型电化学工作站。实验过程中所用溶剂甲苯、N,N二甲基甲酰胺(DMF)、1,2-二氯乙烷(DCE)均经过无水无氧处理的;实验所使用的的玻璃仪器(反应瓶、冷凝管、层析柱等)购自北京欣维尔玻璃仪器公司;柱层析所用硅胶(分析纯)购自从青岛海洋化工有限公司;其他所有试剂若无特别说明,均为市场销售分析纯直接购买使用。
1.2 合成路线
以氟取代的苯并噻二唑为起始原料,通过Still coupling和Knoevengel等反应分别合成了小分子BT2T-IC和端基是氟取代的小分子BT2T-IC4F,具体合成路线如图1所示。化合物M2参照文献[19]报道的方法合成。
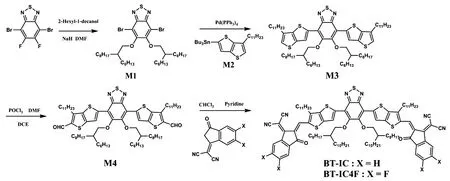
图1 BT2T-IC和BT2T-IC4F的合成路线
1.3 化合物BT2T-IC和BT2T-IC4F的合成
1.3.1 化合物M1的合成与表征
取一个100 mL的两口瓶,抽真空30 min,在氮气保护下加入2-Hexyl-1-decanol(1.84 g, 7.58 mmol),将其溶解在DMF(56 mL)中,在0 ℃下加入氢化钠(0.30 g, 7.58 mmol),搅拌反应1 h。然后加入4,7-二溴-5,6-二氟苯并噻二唑(1.00 g, 3.03 mmol)恢复至室温,搅拌反应12 h。反应结束后,用二氯甲烷萃取,无水硫酸镁干燥有机层,减压旋蒸去溶剂,用n(石油醚)∶n(二氯甲烷)=9∶1硅胶柱层析洗脱,得到产物为无色油状物(1.86 g),产率为79.10%。1H-NMR(400MHz, CDCl3): δ(ppm): 4.12(d,2H),1.91(m, 2H),1.27(m,48H),0.88(m,12H)。
1.3.2 化合物M3的合成与表征
将化合物M1(1.50 g, 1.94 mmol)、化合物M2(3.14 g, 5.82 mmol)和四三苯基膦钯(112.08 mg)加入到50 mL的两口瓶中,加入甲苯(15 mL)将其溶解,氮气保护。油浴110 ℃,冷凝回流反应72 h。反应结束后,冷却至室温,用二氯甲烷萃取,无水硫酸镁干燥有机层,减压旋蒸除掉溶剂,用石油醚和二氯甲烷梯度硅胶柱层析洗脱,得到产物为红色粘稠状液体(1.42 g),产率为82.10%。1H-NMR(400MHz,CDCl3)δ(ppm):4.12(d,2H),1.91(m,2H),1.27(m,48H),0.88(m,12H)。
1.3.3 化合物M4的合成与表征
在50 mL的两口瓶中将化合物M3(1.03 g, 1.20 mmol)溶解在DCE(10 mL)中,氮气保护。再取一个50 mL的两口瓶,加入DMF(0.93 mL,12 mmol),在冰浴下缓慢滴加三氯氧磷(1.68 mL,18 mmol),氮气保护,搅拌反应1 h。将溶解好的化合物M3加入到DMF和三氯氧磷的反应瓶中,氮气保护,反应12 h。反应结束后,冷却至室温,加水萃灭反应,用二氯甲烷萃取,无水硫酸镁干燥有机层,旋蒸除掉溶剂,用n(石油醚)∶n(二氯甲烷)=6∶4硅胶柱层析洗脱,得到产物为红色固体(0.86 g),产率83.53%。1H-NMR(400HZ,CDCl3)δ(ppm):10.14(s,2H),8.58(s,2H),4.02~4.04(d,4H),3.16~3.19(t,4H),2.0(m,2H),1.89(m,4H),1.36(m,66H),0.88(m,18H)。
1.3.4 BT2T-IC的合成与表征
在50 mL的两口瓶中加入化合物M4(150 mg,0.12 mmol),吡啶1 mL,IC(142.15 mg, 0.73 mmol),氯仿15 mL,氮气保护下,70 ℃油浴反应过夜。反应结束后冷却至室温,将反应后的产物逐渐的滴加到甲醇溶液中,让其搅拌沉淀,然后过滤,并依次反复用甲醇、丙酮等溶剂对过滤得到的产物进行多次洗涤,最终得到产物为深蓝色固体(156.96 mg),产率为83.53%。1H-NMR(400 MHz,CDCl3)δ(ppm):9.09(s,2H),8.68~8.70(d,4H),7.96(m,2H),7.77(m,4H),4.11~4.13(d,4H),3.17~3.21(t,4H),2.04~2.11(d,2H),1.84(m,4H),1.25(s,66H),0.86(m,18H)。13C-NMR(400 MHz,CDCl3):δ(ppm):δ=190.43,146.45,144.07,130.34~142.97,137.56,135.89,131.84,130.56,128.42,126.31,121.80,121.83~122.85,116.38,115.92,63.37,31.94,22.23~29.71,14.11。
1.3.5 BT2T-IC4F的合成与表征
在50 mL的两口瓶中加入化合物4(150 mg, 0.12 mmol),吡啶1 mL,IC(142.15 mg, 0.73 2mmol),氯仿15 mL,在氮气保护下,70 ℃油浴反应过夜。反应结束后冷却至室温,将反应后的产物逐渐的滴加到甲醇溶液中,让其搅拌沉淀,然后过滤,并依次反复用甲醇、丙酮等溶剂对过滤得到的产物进行多次洗涤,用n(石油醚):n(二氯甲烷)=1:1硅胶柱层析洗脱,最终得到产物为深蓝色固体(162.01 mg),产率为83.56%。1H-NMR(400 Hz,CDCl3)δ(ppm):9.07(s,2H),8.72(s,2H),8.51~8.55(t,2H),7.70-7.73(t,2H),4.12~4.14(d,4H),3.17(s,4H),2.11(s,2H),1.83(s,4H),1.25(s,66H),0.84(m,18H)。13C-NMR(400 MHz,CDCl3):δ(ppm):δ=190.43,146.45,144.07,130.34~142.97,137.56,135.8,130.56,128.42,126.31,121.83~122.85,116.38,115.92,63.37,31.94,22.23~29.71,14.11。
2 结果与讨论
2.1 小分子化合物的结构分析
图2为小分子BT2T-IC和BT2T-IC4F的核磁共振氢谱图。从图2(a)中分析得出,化学位移在9.09×10-6处所出峰为中心核与端基IC所桥联的双键上氢原子的信号峰,(8.68~8.70)×10-6和(7.76~7.78)×10(-6)出峰为端基IC上苯环上氢原子的信号峰。从图2(b)中分析得出,化学位移在(8.68~8.70)×10(-6)出峰为端基IC4F上苯环上氢原子的信号峰,(7.95~7.91)×10(-6)出峰为并噻吩上噻吩环上氢原子的信号峰,(4.11~4.13)×10-6出峰为烷氧基链上CH2的信号峰,(3.17-3.21)×10-6出峰为并噻吩所连烷基链处CH2的信号峰。对比图a和b小分子BT2T-IC4F在(7.76~7.78)×10-6处的氢原子信号峰消失了,由此分析核磁共振氢谱可得出化合物BT2T-IC和BT2T-IC4F是合成的目标小分子。
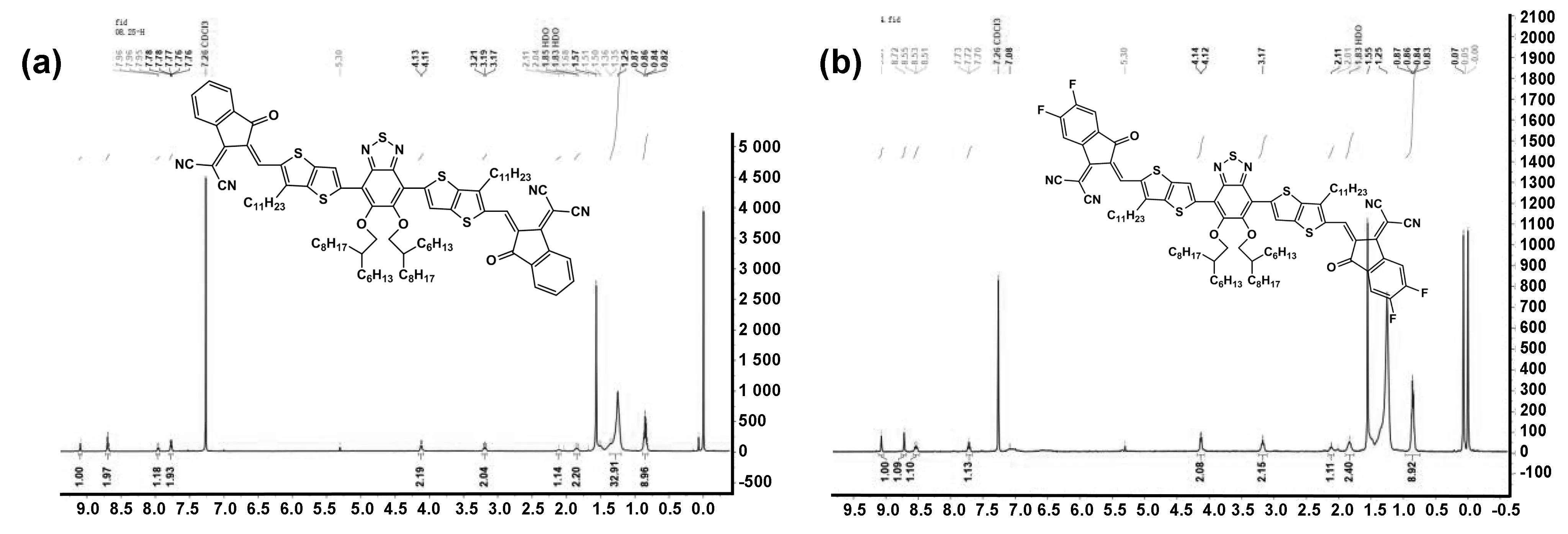
图2 BT-2T-IC 和 BT2T-IC4F的1H NMR谱
2.2 小分子化合物的热力学性质
化合物BT2T-IC和BT2T-IC4F的热力学性质是由热失重(TGA)进行表征测试,在10 ℃/min的升温速率下,测定小分子在氮气中的质量变化,其热失重曲线如图3所示。从图3中可以看出,小分子BT2T-IC和BT2T-IC4F在质量损失为5%时所对应的热分解温度(Td)分别为311.69 和299.06 ℃,表明这两个小分子均具有相对良好的热稳定性。
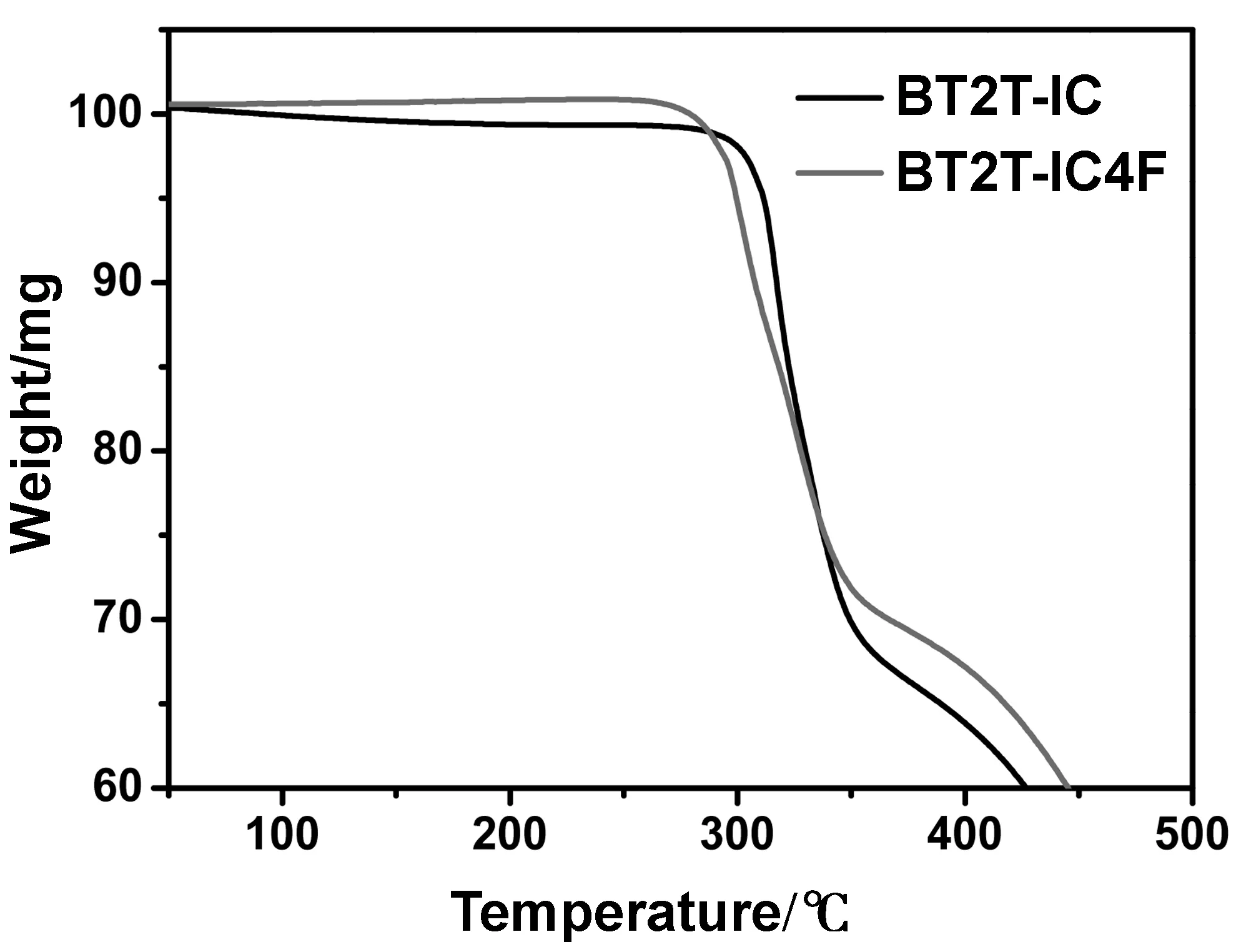
图3 BT2T-IC和化合物BT2T-IC4F的TGA曲线
2.3 小分子化合物的光谱性质
小分子BT2T-IC和BT2T-IC4F在氯仿溶液和薄膜状态下的紫外-可见光吸收光谱如图4所示。从图4(a)可知,在氯仿溶液中,两个小分子在500~700 nm波长范围内均出现一个吸收峰。相对于BT2T-IC,小分子BT2T-IC4F的最大吸收峰略有红移(红移距离9 nm),这可归因于BT2T-IC4F端基上引入电负性更强的F原子,可增强分子的吸电子能力,从而增强分子内电荷转移作用。从图4(c)BT2T-IC和BT2T-IC4F在氯仿溶液中的摩尔吸光系数(ε)分别为1.1×105和1.3×105cm-1,这可能由于BT2T-IC4F中末端中F原子的存在更有利于分子的平面性,从而有利于分子吸收。从两个小分子在固态薄膜状态下的紫外-可见光谱图(图4 b)可知,两个小分子均在661、728 nm左右处出现明显的吸收峰,对应于分子的0-0和0-1的光学跃迁,这分别归因于分子内和分子间的电荷转移效应[20]。同时,相对于溶液态,两个小分子化合物的薄膜态最大吸收峰分别红移了115和102 nm,这可能是由于苯并噻二唑(BT)核上引入的氧原子和并噻吩基团上氢原子,以及并噻吩上的硫原子与酮羰基上氧原子,可通过分子内S…O和O…H作用形成非共价键构象锁从而提高分子的平面骨架堆积,增加分子的有效共轭长度,同时增强分子内和分子间相互作用[20]。同时,化合物BT2T-IC和BT2T-IC4F固体薄膜状态下的吸收起始波长分别在787 和780 nm,其所对应的光学带隙(Egopt)分别为1.57 和1.59 eV,相应的光学数据见表1。
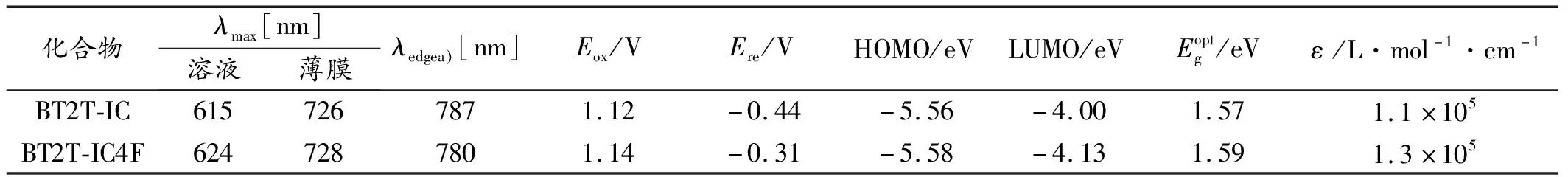
表1 小分子化合物的光学性能和电化学性能参数

图4 BT2T-IC和BT2T-IC4F分别在氯仿溶液(a),薄膜(b)中归一化的紫外-可见吸收光谱,溶液中的吸光系数(c),薄膜中吸光系数(d)
2.4 小分子化合物的电化学性质
通过循环伏安法(cyclic voltammetry,CV)测试了BT2T-IC和BT2T-IC4F的氧化电位和还原电位,然后根据公式(1)和(2)计算出两个小分子化合物的HOMO和LUMO能级。图4为两个小分子化合物BT2T-IC和BT2T-IC4F的循环伏安曲线,由此计算得到的HOMO和LUMO能级数据见表1。由图5可知,BT2T-IC和BT2T-IC4F两个小分子的氧化起始电位基本没有变化,但其还原起始电位差别较大(BT2T-IC的Ere为-0.44 V,BT2T-IC4F的Ere为-0.31 V),从而导致化合物的LUMO能级发生较大变化。这个结果表明端基氟原子的引入基本没有改变分子的HOMO能级,但能有效调节分子LUMO能级,这与文献报道结果是一致的[21]。
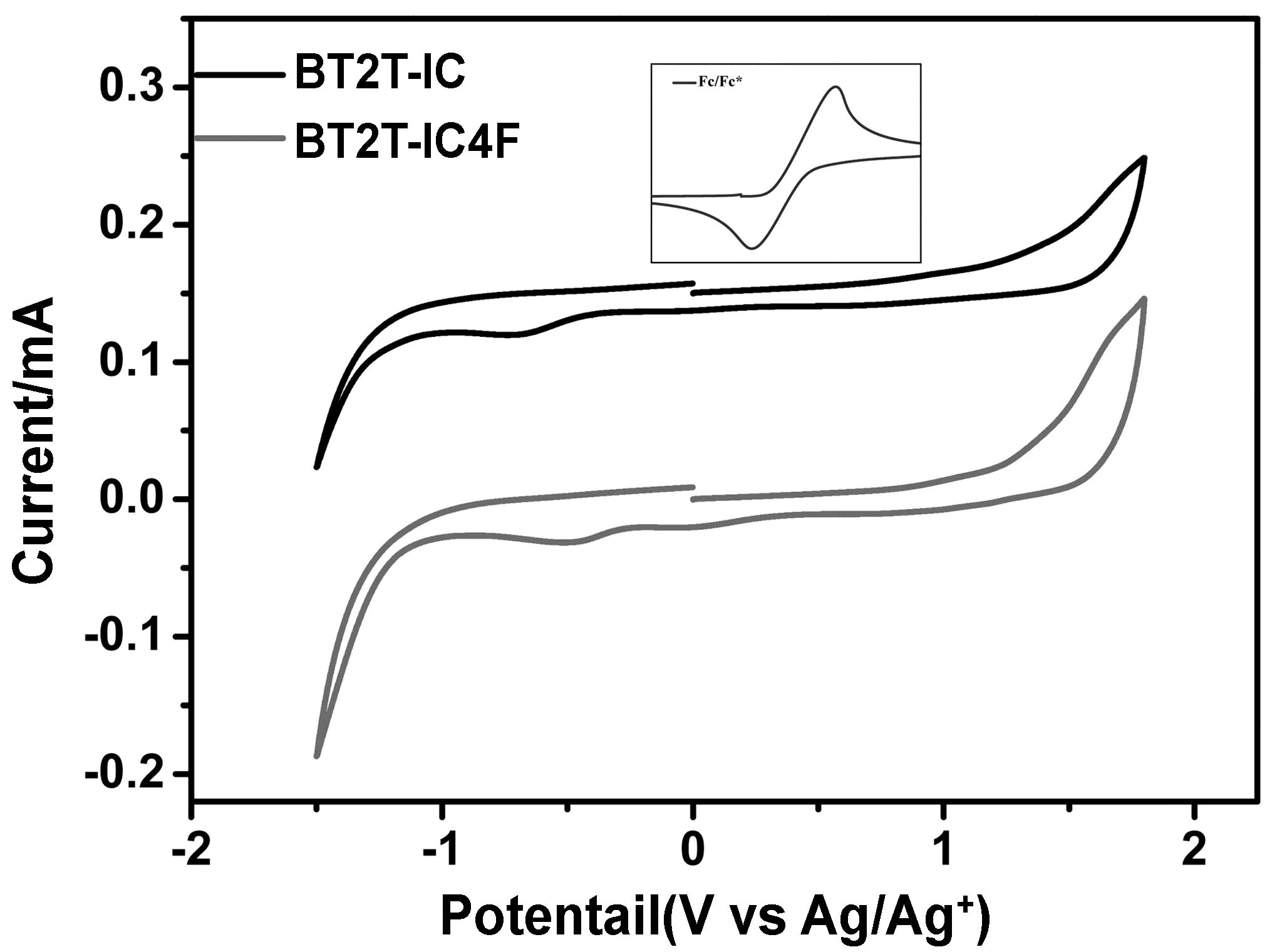
图5 BT2T-IC和BT2T-IC4F的循环伏安测试曲线
EHOMO=-[e(Eox-EFc/Fc+)+4.8]
(1)
ELUMO=-[e(Ered-EFc/Fc+)+4.8]
(2)[21]
2.5 理论计算
BT2T-IC和BT2T-IC2F基态下的几何构型和电子云分布情况通过Gaussian 09软件以B3LYP/6-31G(d)为基组的密度泛函理论(DFT)进行计算。图6给出了其最优基态下的立体结构及HOMO-LUMO级的电子云分布情况并标出了其计算所得的能级值。理论计算的结果显示,由于苯并噻二唑上的烷氧链和桥联并噻吩以及并噻吩和端基IC之间的非共价键相互作用使得两个小分子都呈平面性,说明端基F原子的取代对分子的几何构型影响不大,更加平面的几何构型有利于载流子的传输。两个小分子的HOMO和LUMO能级的电子云都均匀分布在整个分子上,这是由于分子末端连接了强吸电子基团IC和IC4F所引起的。在最优结构下,BT2T-IC 计算所得的HOMO和LUMO能级值分别为-5.72 和-3.72 eV,其理论禁带宽度是2.0 eV。BT2T-IC4F的HOMO 能级、LUMO 能级和理论禁带宽度分别是-5.86、-3.77和2.09 eV。理论计算所得数据与循环伏安法所测数据趋势相吻合,但是理论计算的HOMO与LUMO能级的数据要高于循环伏安法所测数值,这在光伏材料中是很普遍的现象,并不影响对实验结果的分析。

图6 密度泛函理论计算小分子BT2T-IC和BT2T-IC4F的HOMO和LUMO能级的电子云分布图
3 结 论
以烷氧基取代的苯并噻二唑为核,噻吩并噻吩作为π桥,分别采用(二氰基亚甲基)靛酮(IC)、5,6-二氟-3-(二氰基亚甲基)靛酮(IC4F)封端,合成了两个小分子化合物BT2T-IC和BT2T-IC4F。这两个小分子都具有良好的热稳定性;从紫外-可见吸收光谱中可以看出两个小分子化合物在溶液和薄膜状态下的吸收光谱较宽,并且相对于溶液态,两个小分子在薄膜态的吸收都有较大的红移,且薄膜态下的两个小分子都出现了肩峰,表明两个小分子化合物在薄膜态存在较强的相互作用,有较好的堆积。而且通过分子内的S…O 和O…H 作用形成非共价键构象锁,提高了分子骨架结构的平面性,增加分子的有效共轭长度,增强分子间相互作用;在端基上修饰电负性更强的F原子,可增强端基的吸电子能力,从而增强分子电荷转移作用。这项工作为我们进一步研究非共价键稠环小分子化合物提供了一个有效的策略。
猜你喜欢
中学生理科应试(2021年10期)2021-12-07
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
中学生数理化(高中版.高二数学)(2020年6期)2020-07-24
兴义民族师范学院学报(2018年5期)2018-12-18
数理化解题研究(2018年19期)2018-08-15
广州城市职业学院学报(2016年2期)2016-07-25
当代化工研究(2016年1期)2016-03-16
合成化学(2015年10期)2016-01-17
应用化工(2014年9期)2014-08-10
郑州大学学报(理学版)(2014年4期)2014-03-01
