
食管癌预后相关的生物标志物及预后模型的建立①
2021-04-23 03:02封志炜李虎玲王小燕马金凤
新疆医科大学学报 2021年4期
封志炜,李虎玲,王小燕,马金凤,王 凯
(新疆医科大学1基础医学院,2公共卫生学院,3学生处,4医学工程技术学院,乌鲁木齐830011)
食管癌是全球最常见的肿瘤之一,因其预后不良也成为世界第六大癌症死亡原因。2018年根据全球癌症统计,食管癌共造成约50.8万人死亡,占所有癌症死亡的5.3%(诊断出约57.2万例新病例,约占所有癌症的3.2%)[1]。中国食管癌新发477 900例,死亡375 000例,发病率和死亡率分别位列全部恶性肿瘤的第三和第四位,约占全球的一半[2]。全球食管癌患者的总体5年生存率为15%~20%,晚期患者的5年生存率低于15%,早期诊断和准确的预后预测是提高生存率的关键[3]。然而,由于癌症在分子和遗传水平上的异质性,即使患者处于同一阶段并接受相似的治疗,其临床结局和预后也各不相同[4]。因此,肿瘤标志物在食管癌的早期诊断、治疗监测、预后评估等环节中的作用越发重要。大量研究报道已经证实在mRNA、lncRNA、miRNA和蛋白水平表达的差异,对食管癌的发生发展起到的至关重要的作用,为食管癌预后提供潜在的生物标记物[5-8]。但目前,只有少数研究集合mRNA、lncRNA和miRNA对食管癌患者预后的综合评估上。因此,本研究通过整合全球癌症数据库中的食管癌样本的mRNA、lncRNA、miRNA和临床病理特征数据来构建预后分险模型,寻找预后不良食管癌患者的生物标志物,为预测食管癌的预后、治疗决策提供有力的理论依据。
1 材料与方法
1.1数据来源及处理 从肿瘤基因组图谱(the cancer genome atlas,TCGA)数据库下载原始RNA测序数据和临床病理数据。使用官方提供的基因组注释数据库将原始数据注释转化为基因表达矩阵。纳入标准:(1)随访时间不少于30 d的食管癌患者;(2)食管原发肿瘤;(3)必须包含mRNA、lncRNA和miRNA基因表达水平相关数据以供分析;(4)患者个人基本信息、病理信息和随访信息作为可选条件。排除标准:(1)食管继发肿瘤;(2)同时含有其他部位原发肿瘤。最终纳入121个样本数据,包括108例食管癌患者组织标本数据和13例癌旁组织标本数据,共注释了18 729个mRNA,11 056个lncRNA和1 881个miRNA基因表达数据。其中13例癌旁组织标本数据均来自于108例食管癌患者。
1.2差异基因分析 使用R语言“DESeq2”软件包筛选肿瘤组织与正常组织差异表达的mRNA、lncRNA和miRNA,并通过火山图和聚类热图进行可视化。由于mRNA、lncRNA和miRNA计数之间的差距,设置了不同的阈值包括adj P(adj.P.val,adj P)和logFC(log fold change,logFC),来识别差异表达的mRNA、lncRNA和miRNA。mRNA的阈值为:adj P<0.05且|logFC|>2.0;lncRNA的阈值为:adj P<0.05且|logFC|>2.0;miRNA的阈值为:adj P<0.05且|logFC|>1.5。
1.3临床预后模型的建立 将筛选出的差异表达数据进行标准化。应用单变量Cox回归分析其预后意义,并选择P<0.05的指标进行进一步分析。利用R语言glmnet软件包的LASSO算法提取与预后相关的关键mRNA和lncRNA。对上述筛选出与生存相关的mRNA、lncRNA和miRNA分别进行多变量Cox回归分析,并将每个与预后相关生物标记物的标准化表达水平与其对应的回归系数相乘来计算每位患者的风险评分。使用的公式如下:风险评分=β1*基因1(标准化表达)+β2*基因2(标准化表达)+…+βn*基因n(标准化表达),其中β表示每个基因的系数,基因表示基因的标准化表达水平值。使用survminer软件包确定最佳临界值(cut-off value),将患者分为高、低风险组。使用基于对数秩检验的Kaplan-Meier分析比较了高、低风险组的总生存期(OS)。对临床病理特征和3种风险评分进行单变量和多变量Cox回归分析以检测与OS相关的独立预后因素,并使用R语言rms软件包构建与预后相关的风险评分和临床病理数据的列线图模型。
1.4模型内部验证 利用R语言survivalROC软件包绘制ROC曲线以检验模型的区分度,从而减少过度拟合的可能性。通过使用R语言stdca软件包绘制决策曲线以评价模型在临床中的实际应用。使用bootstrap重抽样对模型进行内部验证。
1.5 GSEA富集分析 应用GSEA软件进行基因集富集分析,以识别TCGA队列中高风险和低风险食管癌患者之间的途径和相应的生物标志物的差异,P<0.05的基因被认为是显著富集的。
2 结果
2.1临床特征描述 本研究共纳入108例研究对象,男性89例,女性19例,男女比例为4.7:1。食管癌患者的生存结局中死亡61例,占56.5%,生存47例,占43.5%,其随访时间平均为1.39年。食管癌患者的TNM分期为III期患者41例,I期和II期患者共54例,IV期患者13例。
2.2食管癌差异基因分析 与癌旁组织相比,在食管癌组织中筛选出935个差异表达mRNA,其中456个表达上调,479个表达下调(图1a);筛选出524个差异表达lncRNA,其中331个表达上调,193个表达下调(图1b);筛选出89个差异表达miRNA,其中60个表达上调,29个表达下调(图1c)。筛选出的3种RNA对应的聚类热图分析表明,这些基因在癌组织与正常组织之间呈现差异表达(图2)。
2.3预后相关生物标志物的筛选 通过进行单变量Cox回归和LASSO回归筛选了16种mRNA和10种lncRNA和7种miRNA(图3),纳入多变量Cox回归中,最终筛选出5个mRNA、4个lncRNA和3个miRNA。基于RNA表达量和回归系数,分别计算患者的3种风险评分。根据风险评分的临界值将TCGA队列中的食管癌患者分为高风险组和低风险组,Kaplan-Meie r曲线显示,高风险组的OS比低风险组的OS低(P<0.001),见图4。

图1差异表达的mRNA、lnc RNA和miRNA的火山图

图2差异表达的mRNA、lnc RNA和miRNA的聚类热图
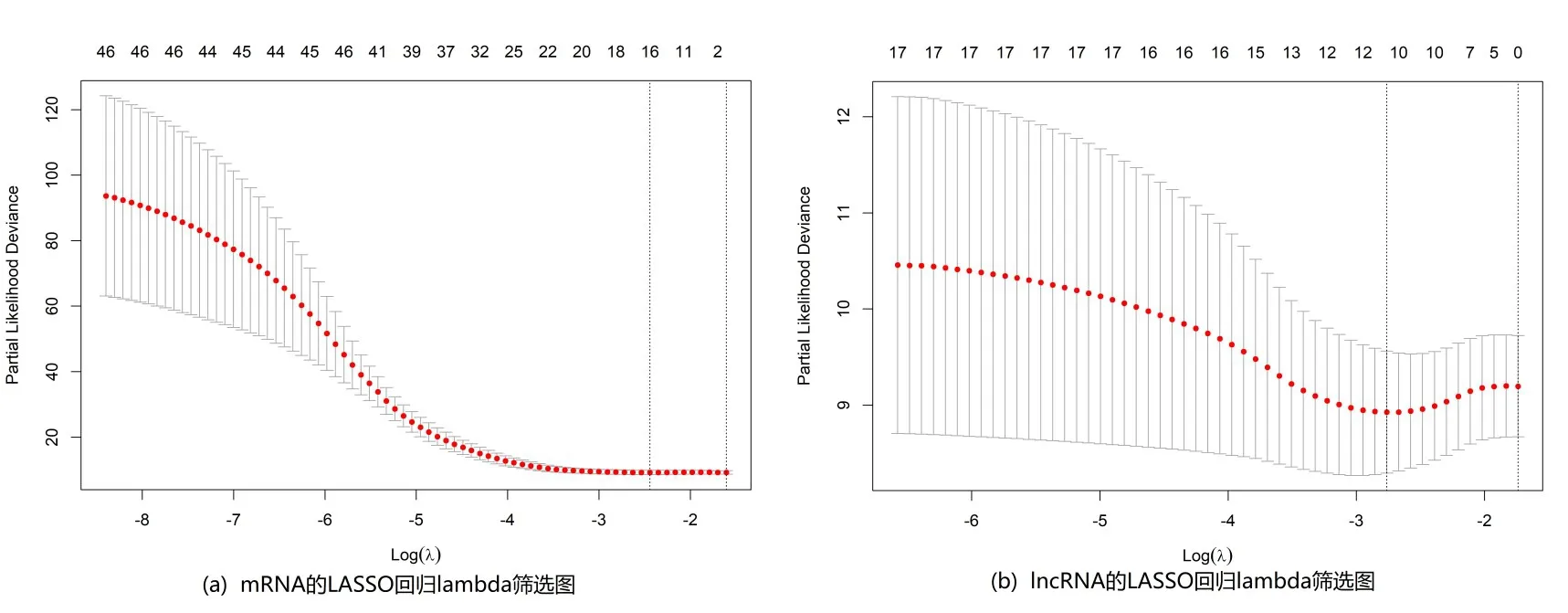
图3 mRNA和lnc RNA的LASSO回归lambda筛选图
2.4基于风险评分预后列线图模型的构建 单变量Cox回归显示,性别、年龄、身高、体重、病理学分型、吸烟和饮酒与食管癌患者的预后无关,mRNA score(HR=2.718,P<0.001)、lncRNA score(HR=2.718,P<0.001)、miRNA score(HR=2.718,P<0.001)和TNM分期(HR=9.591,P=0.005)与食管癌患者的预后相关,如表2所示。多变量Cox回归的结果显示mRNA score(HR=2.121,P<0.001)、lncRNA score(HR=1.938,P<0.001)和miRNA score(HR=1.894,P=0.009)作为连续变量与食管癌患者的总体生存期显著相关。TNM分期IV期患者的预后比I期患者的预后差(HR=6.553,P=0.030)。结 果 表 明mRNA score、lncRNA score和miRNA score是独立于临床特征的预后因子,可以将其作为预后模型的预测指标。预后列线图预测模型显示mRNA score和lncRNA score对预后的贡献最大,其次是miRNA score,最后是TNM分期(图5)。

图4 食管癌患者高低风险生存曲线

表1单变量和多变量Cox比例风险模型

图5 食管癌患者的列线图
2.5列线图模型的验证与校准 基于TNM分期的预后模型AUC为0.653(图6a),mRNA score、lncRNA score、miRNA score和TNM分期的列线图模型AUC为0.815(图6b),表明列线图模型的区分度较TNM分期预后模型更好。2个模型在预测1年生存率时,临床净获益比较,可见列线图模型优于单纯TNM分期模型,凸显了5种mRNA、4种lncRNA和3种miRNA在食管癌发生发展过程中具有重要作用(图6c)。同时使用bootstrap内部验证法对食管癌患者的列线图模型的准确性进行验证,通过校准曲线可以看到,结果显示预测值与实测值基本一致,说明列线图预测模型具有很好一致性(图6d)。
2.6 GSEA通路富集分析 与低风险组的基因相比,高风险组指标在调节胃蛋白酶活性(图7a)、减数分裂(图7b)和细胞基质黏附(图7c)富集分析中更为显著,差异有统计学意义。

图7 GSEA通路富集分析
3 讨论
食管癌是癌症死亡的主要原因,与其晚期诊断治疗密切相关,当前应用比较广泛的预后因素是临床病理特征。但是,由于食管癌遗传的异质性,很难通过其临床病理特征来准确预测预后[9]。因此,寻找新的、有效的早期生物标志物是预测预后、治疗靶标的关键。近年来,随着高通量测序技术的普及以及生物信息学分析的进步,基因数据进入大数据时代。TCGA数据库是获取lncRNA、miRNA或mRNA以及与预后相关临床特征信息的最为全面的数据库,大量研究都通过TCGA数据库展开[10]。目前,大部分的研究关注于单一的转录本,或者是lncRNA和mRNA联合分析,对于lncRNA、miRNA和mRNA联合进行食管癌预后分析的研究很少。
本研究为了进一步阐明食管癌中的lncRNA、miRNA和mRNA差异表达谱,对TCGA数据库中的食管癌数据进行差异分析,筛选出331个表达上调和193个表达下调的lncRNA分子,60个表达上调和29个表达下调的miRNA分子,456个表达上调和479个表达下调的mRNA分子。这些lncRNA、miRNA和mRNA差异表达谱对于进一步认识食管癌发生发展的分子机制提供了新依据。
本研究对食管癌中差异基因进行单因素Cox回归分析,经过筛选一共得到12个与预后相关的RNA,包 括SLC26A9、COX6B2、RP13-672B3.2、RXFP3、OSM,5个mRNA;BLACAT1、CTD-2034I21.2、RP11-60A24.3、RP11-1123I8.1,4个lncRNA;hsa-mir-1269a、hsa-mir-135b、hsa-mir-935,3个miRNA。分别计算其风险评分,并建立了mRNA score、lncRNA score和miRNA score的预后标记。本研究通过ROC和DCA可知,联合模型较TNM分期模型有良好的预测效果,利用bootstrap重抽样的方法进行内部验证,结果显示联合模型具有较好的一致性。说明所得到的12个基因在食管癌预后是比较稳定的,具有良好的区分度和校准度,对于食管癌的预后和治疗都提供了更多的生物标志物的选择。
本研究表明在12个独立预后基因中,BLACAT1的过表达是多种癌症中肿瘤进展和患者预后的有力预测指标。其中BLACAT1在食管癌组织中显著过表达,是食管癌患者预后不良的预测因素。此外,BLACAT1的敲低导致食管癌细胞迁移和侵袭能力下降。已有研究表明,敲低BLACAT1可以减轻EZH2表达,并在蛋白质水平上促进E-cadherin表达[11]。这表明BLACAT1可能调节PRC2的形成,从而促进食管癌进程。hsa-mir-1269a基因与8种癌症富集途径和对细胞功能的下游影响有关[12],其在食管癌组织中表达上调,能够促进食管癌细胞的增殖、迁移和侵袭,被作为了食管癌的预后因子[13]。hsa-mir-135b在恶性肿瘤中起癌基因的作用,它可通过靶向3"UTR来抑制心肌蛋白的表达,并促进细胞的增殖,迁移和侵袭,表明它可能是恶性肿瘤治疗的靶标[14]。SLC26A9是一种在消化系统和呼吸系统中表达的阴离子转运蛋白,已被作为囊性纤维化与CFTR共同定位的潜在靶标[15]。细胞色素C氧化酶亚基6B2(COX6B2)通过增强氧化磷酸化功能(OXPHOS)来驱动肿瘤细胞的转移而不是癌细胞增殖,从理论上讲,COX6B2上调OXPHOS的功能以激活嘌呤能受体途径转移PDAC细胞[16]。抑瘤素M(oncostatin M,OSM)是一种具有多种生物学活性的细胞因子,它能够抑制特定肿瘤细胞的生长,刺激一些正常成纤维细胞的生长,近期研究表明其在食管鳞状细胞癌中OSM已经被证明与预后呈现负相关[17]。松弛素家族肽受体3(RXFP3)的启动子高度甲基化,与肿瘤中的微卫星不稳定性显著相关,且被鉴定为潜在的宫颈癌分子标志物[18]。关于RP13-672B3.2、CTD-2034I21.2、RP11-60A24.3、RP11-1123I8.1和hsa-mir-935在癌症肿瘤中相关研究较少,这些基因在食管癌中的生物学作用可做进一步的研究。
本研究许多问题仍有待解决。首先,本研究仅从TCGA数据库下载数据,有一定局限性。其次,lncRNA-miRNA-mRNA网络在肿瘤的发生和发展中起着重要作用,但研究中未能进行构建网络,进行详细分析。最后,未能对筛选出了预后基因进行实验验证,实验验证可能更有助于了解功能机制,从而有助于了解12种基因用于食管癌预后的功能基础。
综上所述,本研究通过使用TCGA数据库中食管癌患者的数据,建立并验证了mRNA、lncRNA、miRNA和临床病理特征用于食管癌的预后模型。为食管癌的临床治疗和预后提供了大量有价值的信息,后续将在临床标本中进行验证并深入探讨这些指标的生物学功能。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
健康体检与管理(2022年2期)2022-04-15
科教新报(2022年10期)2022-03-27
家庭医药(2022年3期)2022-03-24
现代仪器与医疗(2021年2期)2021-07-21
特别健康·下半月(2020年6期)2020-07-06
婚育与健康(2017年8期)2017-10-27
饮食与健康·下旬刊(2016年9期)2016-05-14
湖南师范大学学报·自然科学版(2014年1期)2014-03-13
