
成人原发性噬血细胞综合征合并EB病毒感染1例
2021-04-21 03:09黄磊郭淑利肖蓬莉王旖旎王慧睿
内科 2021年1期
黄磊 郭淑利 肖蓬莉 王旖旎 王慧睿
1 郑州大学附属洛阳中心医院血液内科,洛阳市 471000;2 北京友谊医院血液内科,北京市 100050
【提要】 噬血细胞综合征(hemophagocytic histiocytosis,HLH)是一组以发热,肝、脾肿大,全血细胞减少以及骨髓、肝、脾、淋巴结组织出现噬血现象为主要临床特征的综合征。HLH分为原发性和继发性两种,多数原发性HLH为家族遗传性基因病,常少儿起病,而成人HLH患者很少考虑为家族性HLH。本文报道了1例成人首诊的家族性HLH,并就其发病过程、诊疗经过、治疗预后进行了总结分析,同时通过对患者及其家属进行基因测序,描绘了家族HLH的UNC13D基因家系图,这对成人原发性HLH的诊治具有重要的参考价值。
噬血细胞综合征(HLH)又称噬血细胞性淋巴组织细胞增多症,是一组以发热,肝、脾肿大,全血细胞减少以及骨髓、肝、脾、淋巴结组织出现噬血现象为主要临床特征的综合征。患者在多种致病因素的作用下,淋巴细胞、单核细胞和吞噬细胞系统异常激活、增殖,分泌大量炎性细胞因子,导致严重甚至致命的炎症反应发生。HLH分为原发性和继发性两大类[1],原发性HLH发病率约为1/50 000,70%~80%的患者在1岁以内发病, 90%以上的患者在2岁以内发病,大多患者有家族史。继发性HLH多由感染(包括细菌、真菌、病毒等,尤其是EB病毒是最主要的诱因)、肿瘤、自身免疫性疾病等诱发,多发生于成年人,患者有明确的原发病,其临床特征与原发性HLH相似。
1 原发性HLH的类型及诊断
原发性HLH包括:(1)家族性HLH,通常发生在婴幼儿;(2)免疫缺陷综合征相关的HLH,如Chediak-Higashi综合征、Griscelli综合征、Hermansky-Pudlak综合征等;(3)EB病毒(EBV)驱动的HLH,如X连锁淋巴组织增生综合征(XLP)。其中最常见的是家族性HLH,它是一种常染色体隐性遗传病分为5个亚型,FHL-1相关缺陷基因及编码蛋白至今仍未确定;FHL-2至FHL-5则分别对应了PRF1、UNC13D、STX11及STXBP2基因及相关编码蛋白[2]。依据HLH-2004诊断标准[3],经分子生物学检查明确存在家族性,或已知有遗传缺陷(包括PRF1、UNC13D、STX11、STXBP2、Rab27a、LYST、SH2D1A、BIRC4、ITK等基因病理性突变),或符合以下8项指标中的5项即可诊断为HLH。(1)持续发热时间>7 d,体温>38.5℃;(2)脾脏肿大(肋缘下≥3 cm);(3)外周血血细胞减少(累及两系或三系):血红蛋白(HGB)<90 g/L,血小板(PLT)<100×109/L、中性粒细胞<1.0×109/L且非骨髓造血功能减低所致;(4)三酰甘油水平升高和(或)纤维蛋白原水平降低:三酰甘油>3 mmol/L或高于同年龄参考值的3个标准差,纤维蛋白原<1.5 g/L或低于同年龄参考值的3个标准差;(5)在骨髓、脾脏或淋巴结内找到噬血细胞;(6)自然杀伤(NK)细胞活性降低或缺如;(7)血清铁蛋白≥500 μg/L;(8)可溶性IL-2受体(sIL-2R或sCD25)水平升高。
2 病例资料
患者男,42岁,于2019年2月20日因“间断发热1周”入院。入院前1周,患者劳累后发热,体温最高39℃,伴畏寒,头痛、头晕,咳嗽、咳白痰,双下肢肌肉疼痛,全身乏力,到我院门诊查血常规示:白细胞5.88×109/L、中性粒细胞39.6%、淋巴细胞50.2%、血红蛋白136 g/L、血小板88×109/L;CRP正常,甲/乙型流感监测结果阴性。门诊行抗病毒治疗效果欠佳,遂住院治疗。患者2年前因“三系减少、脾大”于外院给予脾脏切除术(病理示:淤血脾)治疗,之后彩超检查发现肝脏增大,但未再进行进一步诊治。
入院后复查血常规示白细胞3.67×109/L、中性粒细胞39.6%、淋巴细胞50.2%、血红蛋白108 g/L、血小板63×109/L;实验室检查发现LDH 258 U/L、FER 600 ng/mL;EB病毒抗体阳性,多次检查显示EBV-DNA明显升高;脑脊液检查提示隐球菌感染。腹部CT示肝肿大、肺部感染、胸腔积液、心包积液、腹膜后多发淋巴结肿大。骨髓象示增生活跃,异型淋巴细胞占9%~20%,细胞大小不等,呈不规则形、梭形等。流式免疫表型检测,可见一群表型为CD34-,CD117-,CD2+,cCD3+,CD20-,CD19-,CD5-,CD10-,mCD3-,CD4-,CD8-,CD7-,CD56-,CD16-,CD94-,CD30-细胞;CD45强表达,占有核细胞的9.1%,胞体较大,为异常T淋巴细胞。骨髓活检提示骨髓增生极度活跃、淋巴细胞比例增高;EBV+淋巴细胞增殖性疾病;慢性活动性EB病毒感染(CAEBV)不除外。TCR重排(T细胞受体基因重排)阳性;全身PET-CT扫描阴性。淋巴结活检示淋巴细胞反应性增生;EB病毒原位杂交(+)[2年前脾脏活检标本重新阅片示:窦内大量淋巴组织浸润,并有噬血现象;EB病毒原位杂交散在(+)]。sCD25升高为2 777 U/mL,NK细胞活性减低为3.57%。患者合并EBV感染,根据HLH-2004诊断标准,诊断为EB病毒感染继发的HLH。给予莫西沙星、头孢唑肟抗感染治疗,给予更昔洛韦、帕拉米韦抗病毒治疗,给予两性霉素B、氟胞嘧啶联合氟康唑抗隐球菌治疗,采用VP-16联合地塞米松化疗方案规范治疗。
专家会诊讨论后,考虑到该患者病史较长、长期处于免疫缺陷状态、合并隐球菌感染、多系统受累,拟诊断为隐匿起病的原发性HLH。为此,对该患者进行了HLH相关致病基因套组二代测序,筛查噬血相关基因外显子编码区域突变,结果发现:患者存在UNC13D基因部分位点突变,包括UNC13D基因Exon10上存在无义突变c.766C>T(p.Arg256Ter)(杂合,变异频率51.3%),可能为致病位点(见图1);UNC13D基因Exon6上存在错义突变c.518C>T(p.Thr173Met)(杂合,变异频率50.33%,PolyPhen-2 humVAR值为0.991,SIFT为0.00),临床意义暂不明确(见图2)。据此,患者被确诊为原发性HLH-家族性HLH。经抗感染、抗隐球菌治疗及采用VP-16联合地塞米松化疗方案控制噬血症状后,患者发热、肝脏肿大等症状逐渐好转出院,目前接受门诊治疗、随访,患者口服环孢素维持,血常规基本稳定,LDH、铁蛋白逐渐降低至正常;患者与其大哥配型提示半相合,下一步拟行半相合造血干细胞移植治疗。

图1 患者UNC13D 基因Exon10测序结果
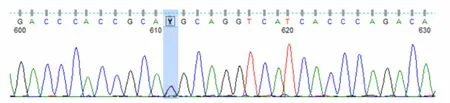
图2 患者UNC13D 基因Exon6测序结果
3 患者的原发性HLH家族史
患者被确诊后,对其家系成员进行了UNC13D基因外显子编码突变筛查发现,可能致病位点来自其母亲(UNC13D基因Exon10上无义突变c.766C>T)(见图3),其二哥[UNC13D基因Exon10上错义变异 c.766C>T(p.Arg256Ter)](见图4)和儿子[UNC13D基因 Exon10上存在无义变异 c.766C>T(p.Arg256Ter)](见图5)也存在该位点突变,但3人均无相关病史。患者父亲未被证实致病的基因变异[UNC13D基因 Exon6上存在错义变异 c.518C>T(p.Thr173Met)](见图6);患者大哥无明显致病基因突变(见图7)。检查结果进一步佐证了患者为家族性HLH(见图8)。
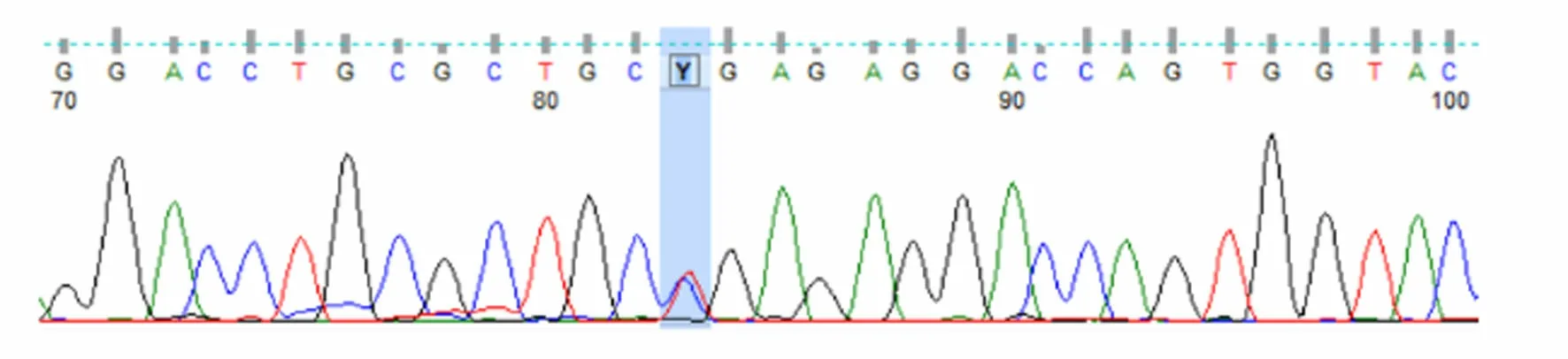
图3 患者母亲UNC13D基因Exon10测序结果
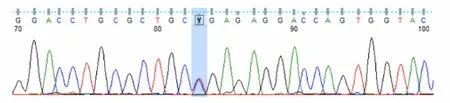
图4 患者二哥UNC13D基因Exon10测序结果
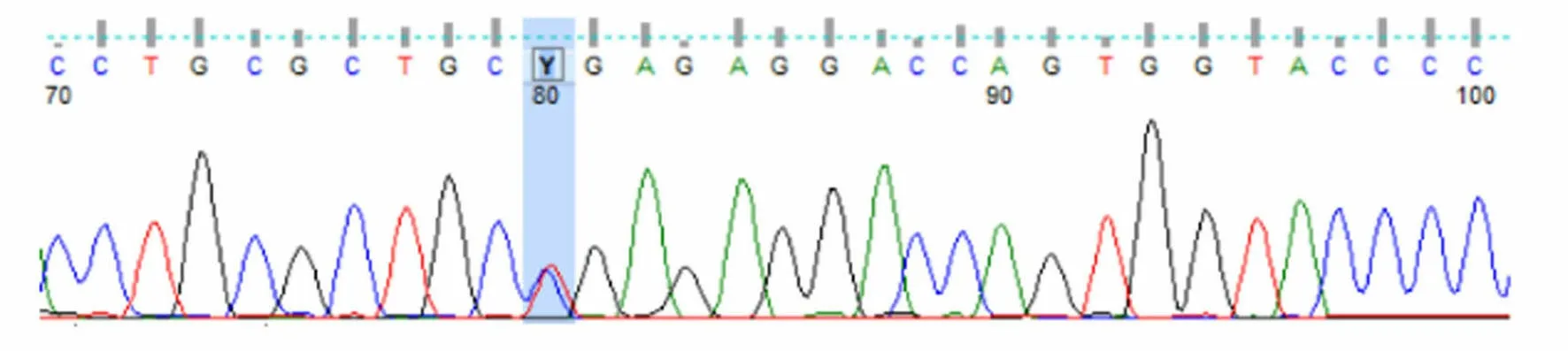
图5 患者儿子UNC13D基因Exon10测序结果
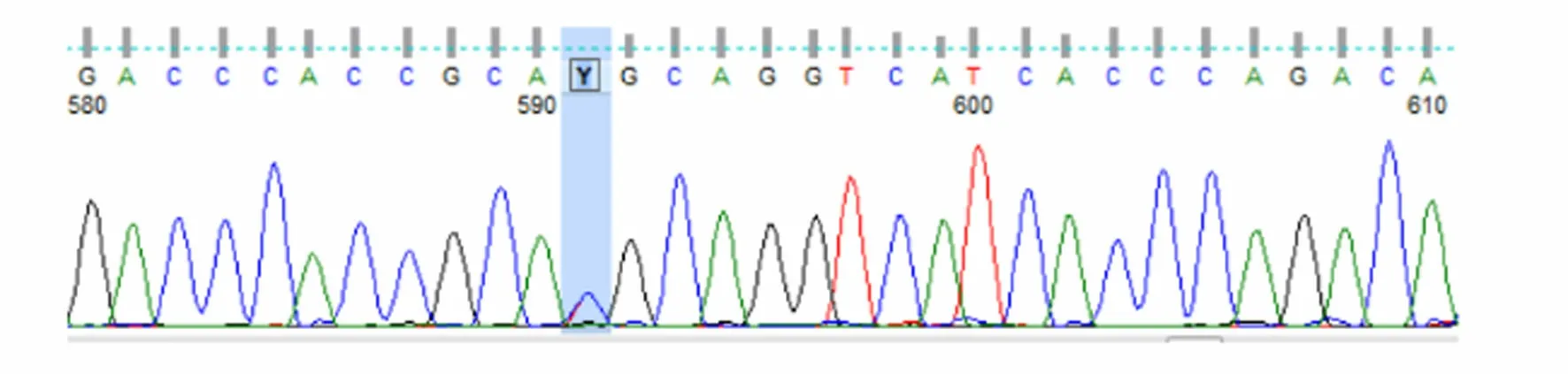
图6 患者父亲UNC13D 基因 Exon6测序结果

图7 患者大哥UNC13D基因外显子测序结果正常
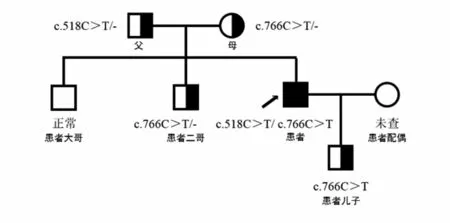
图8 患者家族性HLH的UNC13D基因家系图
4 讨 论
原发性HLH患者多为遗传性HLH,现有报道的患者几乎均为婴幼儿,成人HLH常被诊断为继发性HLH。近年来,随着基因诊断技术的发展,陆续5个家族性HLH相关基因(UNC13D、PRF1、STX11、SH2D1A、RAB27A)突变被鉴定和认可,国际组织细胞协会据此制定了HLH-2004诊断指南,明确把基因缺陷作为诊断和鉴别原发性家族性HLH的标准。Wang等[4-6]相继通过HLH相关基因突变检测确诊了多例成人家族性HLH患者,其中Sieni等报道的11例成人家族性HLH患者的年龄为18~43岁,中位年龄为23岁。
本例患者反复发热、全血细胞减少、肝脾淋巴结肿大、血清铁蛋白水平升高、血清EBV-DNA阳性、合并中枢神经系统隐球菌感染、NK细胞活性明显下降、可溶性CD25升高,根据HLH-2004诊断标准,开始时初步考虑为EB病毒感染继发的HLH。在抗感染、抗隐球菌治疗及化疗后,患者发热、肝脏肿大等症状逐渐好转,血清EBV-DNA转阴,脑脊液检测结果恢复正常;患者病史较长、长期处于免疫缺陷状态并合并隐球菌感染,不排除患者为原发性HLH的可能,故对患者进行HLH相关基因二代测序,结果证实患者存在UNC13D基因突变,最终诊断为原发性HLH合并EB病毒感染。通过对患者家系成员行HLH相关基因测序发现,患者的可能致病位点来自其母亲UNC13D基因 Exon10(存在无义突变),其二哥和儿子也同时存在该位点突变,其父亲在UNC13D基因Exon6上存在错义突变但临床意义暂不明确。据此,最终确诊该患者为家族性HLH。由此可见,对成人HLH患者进行HLH相关基因筛查十分必要。
HLH患者预后差,病死率较高,Rivière 等[7]报道,成人HLH患者的病死率达58%。目前HLH患者的一线治疗方法有HLH-94方案和HLH-04方案,HLH-04方案较HLH-94方案增加了环孢素A(cyclosporine A,CsA)。CsA可降低T细胞活性,减少炎性因子分泌,但CsA肝肾毒性明显。本例患者病情较重,发病时合并神经系统隐球菌、EB病毒感染,CsA应用受限,考虑到HLH-04治疗方案不具有优势[8],因此选用HLH-94方案进行治疗。患者经化疗、抗感染治疗后,病情好转,目前门诊随诊,口服CsA维持治疗;患者体温正常、肝脏体积和肝功能、LDH、铁蛋白正常,病情稳定。由此可见,对确诊的成人家族性HLH患者积极进行免疫治疗及化疗,同时加强抗感染、对症支持治疗,能较好地控制病情发展。
化疗可逆转HLH患者的超炎症状态,造血干细胞移植治疗可恢复HLH患者对病原体正常的免疫反应,移植后5年生存率可达71%,5年无事件生存率可达60%[9]。移植的最佳供者为不携带HLH相关基因全相合同胞,其次为非血缘供者,不携带基因单倍体者亦可[10]。本例患者被确诊为家族性HLH,目前虽病情稳定,但病情可能会随时恶化而危及生命,因此即使只有半相合供者,也强烈推荐其进行移植治疗。在诱导缓解的基础上进行移植治疗,可有效提高移植的成功率和患者的长期存活率[11]。移植治疗原发性HLH的移植相关死亡率高达30%,常因感染、肝静脉闭塞症及非感染性肺炎所致,移植排斥率也较高[12]。本患者大哥未检出噬血相关基因突变,且配型为半相合,故优选其大哥为供者捐献造血干细胞。考虑患者稳定期可能极短,因此积极动员患者尽快行同胞半相合造血干细胞移植治疗,目前正处于准备阶段。
HLH发展迅速,病死率高,临床上对反复发热、肝、脾、淋巴结肿大,全血细胞减少的患者需高度警惕,尽快完善铁蛋白、血脂、NK细胞功能、CD107a等相关检查,明确诊断。对成人HLH仍不能排除家族性HLH的可能,对病因不明确、病史较长或反复发作的HLH患者,需尽快完善基因测序排除家族性HLH可能;对继发性HLH患者可针对病因进行治疗。
猜你喜欢
传染病信息(2022年2期)2022-07-15
中国感染与化疗杂志(2022年3期)2022-06-20
中国药学药品知识仓库(2022年9期)2022-05-23
中国药学药品知识仓库(2022年9期)2022-05-23
锦州医科大学报(2022年2期)2022-05-07
中国典型病例大全(2022年7期)2022-04-22
现代仪器与医疗(2021年4期)2021-11-05
现代临床医学(2021年2期)2021-03-29
特别健康·下半月(2019年7期)2019-07-29
腹腔镜外科杂志(2016年9期)2016-06-01
